Not invited to the party? How to make proprietary ADMET insights more accessible
How do you train robust deep learning models when most of the high-quality data is proprietary? Discover how to enrich multitask ADMET predictions using a public proxy for proprietary learnings.
At OpenADMET, our mission is two-fold: we create, curate, and disseminate high-quality datasets, such as ASAP-Polaris, ExpansionRx, and the ongoing PXR Challenge data, while also rigorously evaluating the state-of-the-art methodologies intended to utilize that data.
In our previous blog post, we analyzed how "zero-shot" public models perform on unseen, high-quality benchmarks like ExpansionRx. The conclusion was daunting: without a significant infusion of new data, these models will lack the chemical "experience" to extrapolate to novel drug discovery problems. This brings us back to the age-old question in data science: where do we get more data?
The source of new data is, of course, extremely important. In predictive modeling, we must avoid prioritizing quantity over quality; we need high-fidelity data from reliable, reproducible assays. While sourcing such data is one of the pillars of OpenADMET's mission, most of the highest-quality labels are typically locked behind the IP barriers of Big Pharma (understandably so). However, inspired by recent work from researchers at Novartis on Targeted Protein Degraders (TPD) [1], we explore another alternative: Surrogate Public Data.
The premise is as follows: if we cannot access the proprietary data itself, can we access its "knowledge" through a proxy? According to this approach, a model trained on a large, high-quality set of proprietary data is used to perform inference on thousands of chemical structures from various public sources [1]. While it is more difficult for companies to make the model itself public due to privacy risks over potential membership inference attacks aimed at revealing the underlying training data [2], the model’s predicted labels can safely serve as a surrogate for the proprietary data. Because these labels originate from a model trained largely on high-fidelity single-source data, they may offer better consistency and larger scale than traditional public repositories like ChEMBL, which are often a patchwork of heterogeneous assays.
In this post, we evaluate whether this surrogate approach actually holds up when tested against real-world benchmarks. Specifically, we look at how models trained on these proxies handle the nuances of different ADMET endpoints.
The caveats of training models from ChEMBL data
Before we dive into the results, we must first address the inherent challenge of curating high-quality training sets from heterogeneous repositories like ChEMBL.
Now more than ever, public datasets offer an invaluable resource for training predictive ADMET models. ChEMBL, the largest repository of bioactive molecules, contains data for over 2.88 million distinct compounds and more than 20 million bioactivity data points [3]. However, because ChEMBL is fundamentally designed around target binding and pharmacology, true ADMET assays make up only a tiny fraction of the database (roughly 3.6% of the total database entries). This means that relative to the size of the repository [3], data points for critical ADMET endpoints like Caco-2 permeability, LogD, and PPB are limited.
Moreover, transforming this massive aggregator into an ML-ready training set is not a simple task. Because ChEMBL draws on thousands of literature sources, it cannot harmonize the protocols and reporting standards used across laboratories. As noted by Landrum et al., combining results from such varied assays is notoriously problematic due to incompatible experimental conditions and incomplete assay metadata [4].
To illustrate these implications, we look at the curation process we followed for our in-house lipophilicity and permeability model (available on Hugging Face). Preparing an ML-ready training set for this model involved a multi-stage process including keyword-based searches to harmonize assay descriptions, standardizing units, and applying physically-grounded filters to remove "impossible" outliers. This only scratches the surface of how detailed you can get with data curation and harmonization. Inductive Bio provides an interesting example in their blog, where they discuss how some Human Liver Microsome (HLM) assays in ChEMBL lack a critical cofactor.
Public data curation for lipophilicity and permeability endpoints
Although entries in ChEMBL are manually curated, significant inconsistency remains due to data being submitted from thousands of sources under different assay conditions. Below, we discuss the specific issues encountered during "standard" curation of our training sets.
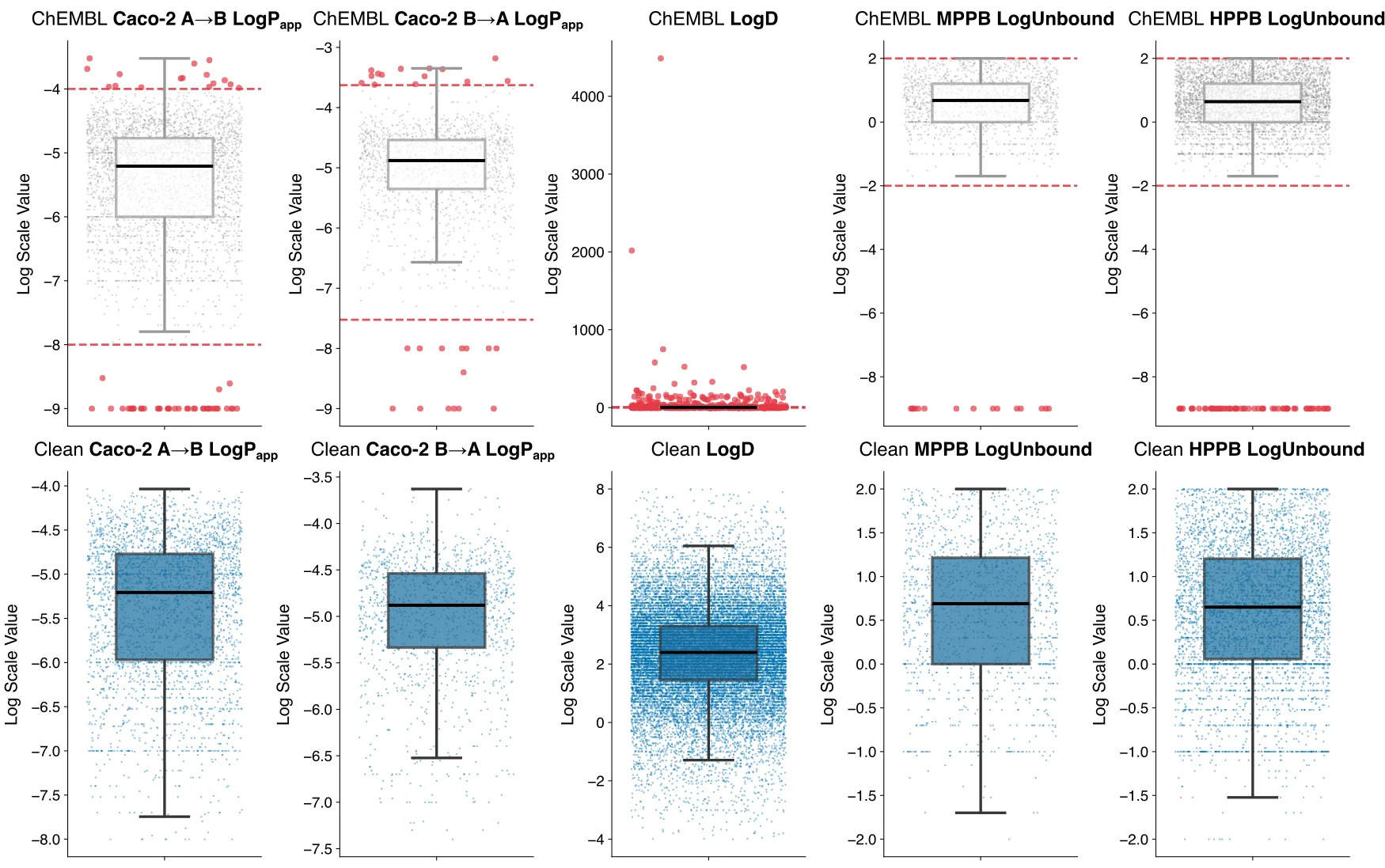
-
Caco-2 permeability
Curation of the Caco-2 dataset was particularly non-trivial due to the lack of standardized nomenclature in assay descriptions. For instance, while the majority of entries for A→B and B→A flux were explicitly labeled as “apical to basolateral” or “basolateral to apical”, a significant subset (248 entries) utilized the keyword “basal” to describe the receiver or donor compartment. Failing to account for these linguistic variations would lead to a substantial loss of valuable training data. Similarly, a number of entries (1158 points) provided only a generic label of “apparent permeability” without specifying the flux direction. While many of these likely represent A→B transport, as this is the standard orientation for absorption studies, the lack of metadata introduces unacceptable uncertainty, especially since the difference between A→B and B→A is used to calculate downstream quantities such as efflux ratio. To maintain high data integrity and avoid injecting directional noise into the training set, we decided to exclude these ambiguous entries.Beyond nomenclature, the assay's pH represents a significant, often hidden, source of experimental noise. Standard Caco-2 protocols typically employ a pH gradient (pH 6.5 Apical / 7.4 Basolateral) to mimic the physiological transition from the acidic intestinal lumen to the neutral environment of the plasma [5]. Because permeability is a function of a drug’s ionization state, even minor pH fluctuations can drastically alter the protonation of acidic or basic molecules. Consequently, consistent pH conditions are a prerequisite for a high-fidelity ML training set. However, our analysis of the ChEMBL records revealed two major complications for pH-based curation:
-
Less than 10% of the total Caco-2 entries explicitly report the assay pH. Performing a 'naive' curation is nearly impossible without resorting to advanced LLM-based methods to extract these details from the original source material.
-
Variation in assay conditions: Approximately 7% of the reported entries were conducted under “iso-pH” conditions (symmetrical pH 7.4), while a small subset of 38 entries showed deviations of up to ±3 units from standard pH.
While these variations, especially the absence of a pH gradient, add biological complexity, the scarcity of metadata makes systematic filtering infeasible. Therefore, we did not perform explicit pH-based curation for this model, but we acknowledge this as a potential source of model underperformance, as the “noise” in the training set likely reflects inconsistent experimental conditions.
The distribution of Caco-2 Papp A→B is shown in Fig. 1. On the top panel, we plot the distribution of these two endpoints as curated from ChEMBL.There is a concentration of points at exactly log Papp=-9 Log(cm/s). This value corresponds to an imputation floor introduced during data curation to handle zero values (avoiding log(0) = -inf). In practice, drugs with extremely low human absorption (<1%) typically report Papp values around 1 × 10⁻⁷ cm/s; measurements below this threshold often fall outside the assay’s limit of detection [6].
Conversely, we observe several points exceeding log Papp > -4 (100 × 10⁻⁶ cm/s). While drugs with complete human absorption generally show Papp > 1 × 10⁻⁶, measured permeability is physically capped by the Aqueous Boundary Layer (ABL). Because the rate of aqueous diffusion limits transport in this layer, values >100 × 10⁻⁶ cm/s are biologically and physically unlikely. Based on this criterion, we filtered out all entries falling outside a fixed physiological range of [-8, -4] log (cm/s), aligning with reported literature standards [7]. Note that because this data is used for multitask training, the labels are dropped at the column level (i.e., set to NaN), to avoid removing entire rows where clean data is available for other endpoints.
In contrast, Caco-2 B→A permeability is skewed toward higher values. This is expected, as B→A flux is often accelerated by active efflux transporters such as P-glycoprotein [6]. At the same time, the ChEMBL distribution for B→A appears less noisy, with fewer points clustered at the floor. Here, the 1st and 99th percentiles (about -7.2 and -3.6 log units, respectively) align well with assay limits, so we apply the same column-level filtering strategy across that specific range.
-
LogD
The raw ChEMBL data for LogD is remarkably noisy (Fig. 1, top panel), containing unphysical outliers with values exceeding 10³ log units! These extremes are likely not measurements at all, but rather artifacts of automated data extraction. To stabilize the distribution for machine learning, we filtered out logD values outside the physically plausible range [-4, 8], which reflects the analytical detection limits of standard lipophilicity assays, following the same column-level strategy used for Caco-2 Papp. This cleaning step results in the more uniform, usable distribution shown in the bottom panel.A secondary challenge in curating this endpoint is the potential 'leakage' of computed or predicted values into the experimental database. While our curation strictly targeted entries labeled “LogD”, public repositories occasionally include values calculated by software and deposited alongside experimental results. Because these computational estimates are not always explicitly flagged in the metadata, they can introduce algorithmic bias into the training set. An example of this occurs when literature publications list software-calculated properties in the same tables as real assay results. In such cases, ChEMBL automated curation pipelines may easily clump these values together and inadvertently deposit the theoretical values into experimental keywords. For example, the ChEMBL assay record CHEMBL839834, contains data labeled explicitly as “Calculated partition coefficient (clogD)”. We acknowledge that without an exhaustive, manual review of the original source literature for every entry, some of these predicted values likely remain in the final dataset, representing a known source of uncertainty in high-throughput ADME modeling.
-
Protein binding
For the Protein Plasma Binding (PPB) dataset, curation was performed by species (e.g., human vs. mouse) to account for inter-species differences in protein affinity. We converted raw % bound values into % unbound prior to performing the logarithmic transformation, to match the values in the ExpansionRx dataset. In the raw distribution (Fig. 1, top), a significant cluster of points at -9 log units represents an artificial floor used during the initial log-conversion for compounds reported with nearly 100% binding. To resolve this, we apply a physically defined cutoff of [-2, 2] log units. This range covers the entire physiological spectrum: the upper bound of +2 represents compounds that are 100% unbound (log10(100) = 2), while the lower bound of -2 corresponds to 0.01% unbound. This lower cutoff acknowledges the analytical limits of standard equilibrium dialysis assays, where differentiating between 99.99% and 100% binding is often beyond practical precision. This approach results in the cleaner, more physically representative distribution shown in the bottom panel.
Even after careful curation, pre-trained models can’t achieve reasonable performance for all endpoints
Ultimately, these curation efforts represent the ceiling of what can be achieved with public data. Despite applying rigorous physical filters and nomenclature harmonization, models trained strictly on public data still struggle when facing complex assays like Caco-2 permeability, often resulting in negative R² values and high absolute errors. This underperformance isn't a failure of the architecture; it is the consequence of the heterogeneity of public data, as we saw during the ExpansionRx challenge.
This raises an important question. If public data isn’t enough (and we, simple mortals, don’t have access to clean, high-quality proprietary data), could ML-predicted surrogate data provide a proxy to more homogeneous data sources? In the next section, we examine how the models perform when we introduce these surrogate data into the mix.
Training Surrogate models
Recent efforts led by Novartis may offer an alternative to increasing public data availability, while keeping proprietary molecules private [1, 8]. To achieve this, the team designed a two-step knowledge-distillation pipeline (illustrated in the top panel of Figure 2) and applied it to Targeted Protein Degraders (TPDs), a highly promising but data-scarce therapeutic modality. First, they constructed a highly accurate multitask (MT) model by ensembling message-passing neural networks (MPNN) and a feed-forward deep neural network (DNN), training it on thousands of internal, historical data points. Then, they used this high-fidelity “Oracle” model to perform inference on a massive library of over 274,000 compounds sourced from public datasets like ChEMBL, ZINC, and PROTAC-DB. By doing so, they generated an entirely open-sourced surrogate dataset annotated with ML-predicted labels for 25 key molecular endpoints.
Importantly, the study found that models trained on this surrogate data performed comparably to the original proprietary models on key endpoints on TPD molecular glue assays. While the Novartis results suggest that surrogate models can successfully capture the behavior of these complex degradation endpoints, they also noted performance gaps where the surrogate chemical space didn't fully align with the molecules being predicted.
This leads us to a critical question: how do these surrogate models perform when evaluated on a well-defined, unseen chemical space such as the ExpansionRx dataset?
Evaluating the performance of surrogate models on a focused dataset
The Novartis surrogate dataset is the product of four large-scale MT-GNN models across 25 endpoints, designed to capture the interplay between related ADME properties. These models include:
- Permeability (5-task): Covering Papp from LE-MDCK, PAMPA, and Caco-2, alongside efflux ratios from MDCK-MDR1.
- Clearance (6-task): CLint across a broad species spectrum (Rat, Human, Mouse, Dog, Cyno, and Minipig).
- Binding/Lipophilicity (10-task): Including PPB for five species, HSA binding, microsomal/brain binding, and LogP/LogD.
- CYP Inhibition (4-task): Covering reversible and time-dependent inhibition of major isoforms.
To evaluate the applicability of models trained on these surrogate labels (hereafter, surrogate models) to the ExpansionRx dataset, we focused on the endpoints that overlap between the two datasets (Novartis and ExpansionRx).
- For the binding/Lipophilicity model, we evaluated mouse plasma protein binding (MPPB), mouse brain binding (MBPB), and LogD.
- From the permeability model, we focus on Caco-2 permeability (Papp) A→B.
- From the clearance model, we focus on human and mouse intrinsic clearance, HLM CLint, and MLM CLint.
For each of these task groups (each trained as a MT-GNN), we want to answer the following questions: Can surrogate models effectively extrapolate to unseen space? And, can pre-training with these models improve performance compared to public data (e.g., ChEMBL). For this purpose, we train CheMeleon [9] pre-trained models using five different strategies, and perform inference on the blinded ExpansionRx test set, using the hyperparameters optimized by the OpenADMET team (see Figure 2):
- Base model: CheMeleon MT trained on the ExpansionRx training data for each task group.
- Surrogate extrapolated model: CheMeleon MT trained on the surrogate data.
- Surrogate fine-tuned model: CheMeleon MT pre-trained on the surrogate data and fine-tuned with the ExpansionRx training data.
- ChEMBL extrapolated model: CheMeleon MT pre-trained on ChEMBL data.
- ChEMBL fine-tuned model: CheMeleon MT pre-trained on ChEMBL data and fine-tuned with the ExpansionRx training data.
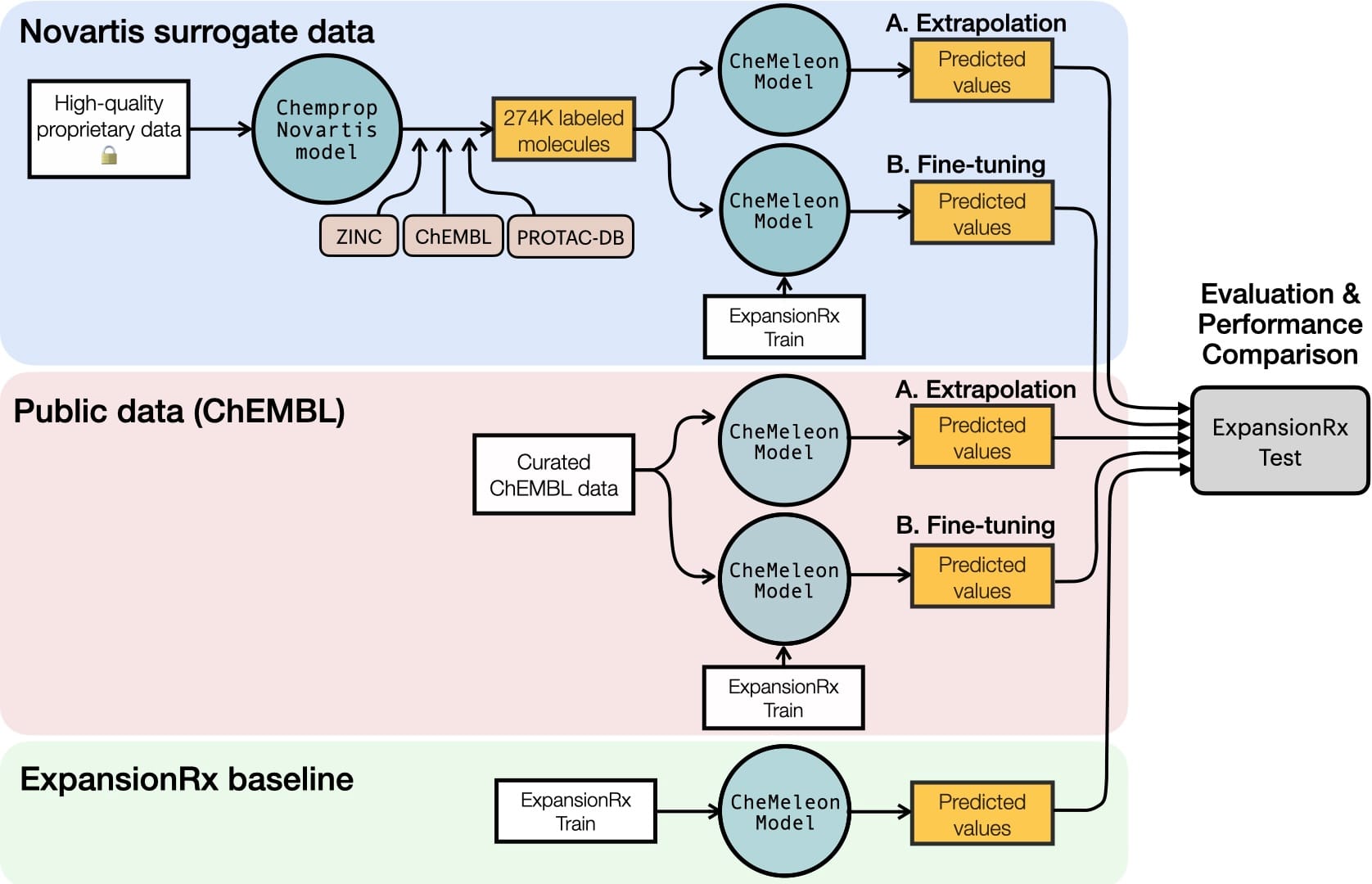
We chose to present results using the foundational CheMeleon architecture to maintain a consistent framework across our different data strategies, as this matches our in-house ChEMBL pre-trained models. To ensure these findings were robust, we benchmarked the same experiments using a Chemprop architecture with the hyperparameters optimized by Peteani et. al. [1], which yielded similar performance trends.
Can surrogate models improve endpoint prediction?
To test whether pre-training with surrogate data can improve endpoint predictions on unseen chemical space, we evaluated and compared the five model training strategies outlined in our workflow (Figure 2) across our three core endpoint task groups.
Global performance across task groups
In the following sections, we evaluate model performance on the blinded ExpansionRx test set using three different metrics: coefficient of determination (R²), absolute error (Mean Absolute Error, MAE), and ranking performance (Spearman’s rank correlation, ρ). Evaluating ranking order is highly relevant for hit prioritization in early-stage discovery, where identifying the relative trend of a chemical library is often more critical than predicting an absolute numeric value.
To ensure statistical rigor, performance metrics and confidence intervals were calculated using 1,000 bootstrap samples of the test set. Bootstrapping was chosen instead of the standard cross-validation procedures often recommended in the literature [10], because it allows a direct statistical comparison between our locally trained models and the extrapolation models. As recommended in our previous blog post, we choose to present these results using pairwise differences (Δ) of each model relative to the top-performing model (centered at 0). Because the confidence intervals are derived from the paired bootstrap difference distribution, a model's performance shift is only statistically significant if its error bar does not cross the zero baseline. The absolute benchmark metrics for each top-performing reference model are provided directly within the panel titles for context.
Lipophilicity & Binding
The overall performance profile for the lipophilicity and binding endpoints is summarized in Figure 3.

The results above reveal an important insight: straightforward physical chemistry endpoints benefit the most from localized training. Across all three lipophilicity/binding endpoints, the local base model consistently achieved the best overall performance with the lowest absolute error (MAE) and the highest coefficient of determination (R²), sitting at the reference =0 line in Figure 3. The reason is that lipophilicity and plasma/brain protein binding are heavily driven by physical properties such as hydrophobicity and molecular weight. These properties can be easily learned by the MPPN architecture when trained directly on local, tightly controlled project data.
However, when we look at the fine-tuned strategies, we see a clear case of negative transfer: pre-training on large, heterogeneous chemical spaces (such as ChEMBL and Novartis’ surrogate data) forces the model weights to accommodate a wide variety of assay conditions and diverse chemistry. Then, when we attempt to fine-tune the model on the ExpansionRx dataset, the architecture struggles to completely unlearn these conflicting representations, leading to inflated absolute errors (MAE) and worse regression fits R², when compared to the baseline. This behavior is more clearly observed in MPPB, where adding external data causes the error of all pre-trained models to increase significantly (MAE>+0.2), and results in a severe performance drop in predictive variance (with ChEMBL fine-tuning plunging to a negative absolute score (R² < -1.2) with respect to the R²=0.553 baseline).
Interestingly, the surrogate fine-tuned model still managed to improve the local baseline's ranking performance for both LogD and Log MBPB. Furthermore, for Log MPPB ranking performance, the surrogate fine-tuned model is not statistically different from the baseline, as indicated by its grey error bar crossing the zero line. This seemingly odd behavior, where external pre-training degrades absolute error metrics but preserves or improves ranking capability, strongly suggests a systematic offset between the historical source assays behind the surrogate data and the local ExpansionRx dataset. In other words, the surrogate data is shifting all predictions up or down by a consistent mathematical margin. Because ranking metrics are typically robust against uniform calibration offsets, the model is able to successfully leverage the foundational structural understanding embedded within the surrogate data for compound prioritization.
Generally, we also note that for LogD and MPPB, the surrogate datasets don’t show a clear advantage compared to public data in both pre-training and extrapolation (in fact, for LogD the fine-tuned ChEMBL model is only slightly worse than the baseline), as these endpoints are well represented in public data. For Mouse Brain Binding (MBPB), we report only the baseline and surrogate fine-tuned models, as our in-house model doesn’t currently predict this endpoint.
Permeability
While local training proved superior for straightforward physical properties such as lipophilicity, for more complex biological endpoints such as Caco-2 cell permeability, the results tell a different story (Figure 4).

For this endpoint, the surrogate fine-tuned model is now the best performing model across all metrics, at =0, significantly outperforming the local baseline. Strikingly, even the extrapolated surrogate model sits close to the zero line, performing nearly on par with the baseline trained directly on project data.
In contrast, both fine-tuned and extrapolated models pre-trained on ChEMBL data fall significantly below the zero line, underperforming even relative to the baseline model. Cell permeability is notoriously difficult to harmonize across public data due to differences in cell passages, pH, and experimental conditions between labs (as explored above). These results confirm that highly heterogeneous public data can actually inject statistical noise into the foundational weights of the model, affecting its performance. Novartis’ surrogate data, by contrast, serves as a clean, harmonized scaffold that enables the model to learn the underlying biological mechanism more effectively than local or public data alone.
Clearance
Clearance (CLint) remains an incredibly challenging endpoint for traditional machine learning approaches. It is generally heavily dictated by three-dimensional molecular orientation, steric hindrance at metabolically liable sites, and is highly sensitive to local chemical structure.
Using our standard CheMeleon architecture, fine-tuning with surrogate data primarily helped boost the model's ranking capability, though it came at a slight cost to absolute error metrics such as MAE and R². It is worth noting, however, that superior ranking performance is often the most vital asset in real-world early-stage drug discovery programs, where the primary goal is to prioritize the top compounds for synthesis rather than to predict absolute metabolic values.
Globally, we did not observe a clear trend for clearance, making definitive conclusions difficult. For HLM CLint, pre-training adversely affected the prediction of clearance on the ExpansionRx test set, leaving the baseline CheMeleon model the most stable predictor for absolute values, while, for MLM, the surrogate extrapolated model unexpectedly performs the best in terms of absolute error, sitting at the zero line for MAE and R².
A deep dive into the potential of surrogate models
Extrapolation to unseen chemical space
It is crucial to clarify that surrogate data is not a magic wand that makes models universally better at zero-shot extrapolation compared to public repositories. Success depends heavily on the endpoint architecture. In Figure 6, we compare the regression plots of the predicted values of Caco-2 Papp and LogD in the ExpansionRx test set using the ChEMBL and surrogate-extrapolated models.

With endpoints that are poorly represented in public datasets, such as Caco-2 Papp, the model benefits significantly from surrogate pre-training. While ChEMBL permeability data is both scarce and heterogeneous (as discussed above), the surrogate data employed here (though generated via ML predictions rather than experimental assays per se) is supported by tightly controlled, harmonized assays from industrial drug discovery pipelines conducted under standardized conditions.
For Caco-2, this clean "synthetic anchor" allowed for a boost in the model’s ranking capabilities: Spearman’s ρ jumped from 0.34 to 0.70, and Kendall’s τ rose from 0.23 to 0.51. Similarly, the model’s R² transitioned from -0.01(i.e., equivalent to predicting the mean of the test data every time) up to a borderline acceptable 0.28, all while keeping absolute error (MAE) relatively stable.
In contrast, for endpoints that are mature and well-represented in public data, such as LogD, changing the pre-training source to a surrogate dataset yielded negligible benefits (e.g., Spearman’s ρ changed from 0.69 to 0.71 and MAE from 0.63 to 0.55). In fact, we previously showed (blog post) that even public data extrapolation performs reasonably well for this endpoint.
While we remain cautious about claiming surrogate data provides the ultimate answer to the challenge of zero-shot domain extrapolation, the real gain lies in how these models behave once they are allowed to adapt, as we will show in the next section.
Surrogate models could be an alternative to pre-training with public data
We believe that the biggest impact of surrogate datasets will be their ability to serve as high-fidelity scaffolds for foundational ADMET models before they undergo project-specific fine-tuning. Pre-training an ML architecture on low-quality, noisy data doesn't just give you a poor starting point; it misaligns the base model weights, establishing a poor baseline that’s hard to fix with local-data training.

As shown in Figure 7, external pre-training did not universally guarantee an advantage, and in some cases, led to significant underperformance relative to the simple baseline model. This effect varies with task complexity. For a straightforward physical property like MPPB, where the small dynamic range is already well captured by the baseline model (MAE=0.20 and R²=0.56), global pre-training introduces a performance penalty. For both public and surrogate data pre-training, the MAE doubles to over 0.42, while R² becomes negative (< -0.63).
The true power of surrogate pre-training becomes evident when we look at the complex biological endpoint, Caco-2 Permeability. Here, where public data is notoriously fractured and unharmonized, the baseline model alone struggles. When pre-trained on the harmonized surrogate dataset, the model achieved the best performance across all Caco-2 Papp models, optimizing both absolute precision (MAE = 0.19) and ranking power (Spearman’s ρ = 0.75), whereas ChEMBL pre-training performed worse than the baseline.
But does it actually make a difference on the leaderboard?
To translate these statistical metrics into a real-world context, we mapped our final model performances directly against the final leaderboard of the OpenADMET-ExpansionRx blind challenge.
Table 1. Ranking of all five models on the OpenADMET-ExpansionRx final leaderboard.
| Endpoint | Baseline rank | ChEMBL extrapolation | Surrogate extrapolation | ChEMBL fine-tuned | Surrogate fine-tuned |
|---|---|---|---|---|---|
| LogD | 67 | 97 | 88 | 72 | 95 |
| MPPB | 33 | 67 | 75 | 100 | 100 |
| Caco-2 Papp | 78 | 98 | 87 | 96 | 17 |
Looking at the leaderboard rankings confirms our previous observation: more data does not always equal a better model. In fact, for both LogD and MPPB, attempting to incorporate external data (either for extrapolation or fine-tuning) consistently damaged our leaderboard standings compared to the local baseline model. For Caco-2 permeability, however, we can see the performance improvement is reflected in massive leaderboard gain, with the Surrogate fine-tuned model reaching rank 17 (compared to the baseline's 78).
As noted in our previous blog post, extrapolating models trained on public data, while tempting, is not recommended; doing so here resulted in bottom-tier rankings of 97, 67, and 98, respectively. The variation in performance trends across endpoints serves as a reminder that tailoring the data integration strategy to the specific endpoint should always be the strategy of choice when training ADMET models.
Conclusions
If this benchmark shows anything, it is that there isn't a single “one-size-fits-all” strategy for ADMET modeling. Every endpoint is different and requires a different data setup.
For simple endpoints like lipophilicity or protein binding, local training data performs best. Forcing the model through global pre-training loops may introduce unnecessary noise and undermine absolute predictions. But for complex biology like Caco-2 permeability, where public data is also disjointed and messy and local datasets don't have enough breadth to teach the model the underlying mechanics, surrogate data works incredibly well, particularly for ranking and prioritization.
Ultimately, we aren't trying to argue that ML-predicted surrogate data will replace real experimental assays. High-quality experimental data will always be better for model training, but our results highlight the potential of surrogate datasets as a practical “middle ground”.
The results here also highlight that it is the fidelity and consistency of data that matter, not just mere volume. This helps validate our central strategy at OpenADMET: collecting large amounts of homogeneous data from a few high-quality assays run at a single institution.
Rather than waiting for pharma companies to unblind and release proprietary structures, which rarely happens, sharing this kind of distilled proxy data is a realistic way to lift the performance baseline for open-source ADMET tools, and we encourage our partners to do so! In the meantime, we will keep doing our part to get more clean data out into the community by running and releasing quality assays and structures through our upcoming blind challenges.
References
- Peteani, G., Huynh, M. T. D., Gerebtzoff, G., & Rodríguez-Pérez, R. (2024). Application of machine learning models for property prediction to targeted protein degraders. Nature Communications, 15(1), 5764. ↑
- Krüger, F. P., Östman, J., Mervin, L., Tetko, I. V., & Engkvist, O. (2025). Publishing neural networks in drug discovery might compromise training data privacy. Journal of Cheminformatics, 17(1), 38. ↑
- Zdrazil, B., Felix, E., Hunter, F., Manners, E. J., Blackshaw, J., Corbett, S., ... & Leach, A. R. (2024). The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Research, 52(D1), D1180-D1192. ↑
- Landrum, G. A., & Riniker, S. (2024). Combining IC50 or Ki values from different sources is a source of significant noise. Journal of Chemical Information and Modeling, 64(5), 1560-1567. ↑
- Volpe, D. A. (2008). Variability in Caco-2 and MDCK cell-based intestinal permeability assays. Journal of Pharmaceutical Sciences, 97(2), 712-725. ↑
- Artursson, P., & Karlsson, J. (1991). Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochemical and Biophysical Research Communications, 175(3), 880-885. ↑
- Hubatsch, I., Ragnarsson, E. G., & Artursson, P. (2007). Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nature Protocols, 2(9), 2111-2119. ↑
- Fluetsch, A., Trunzer, M., Gerebtzoff, G., & Rodriguez-Perez, R. (2024). Deep learning models compared to experimental variability for the prediction of CYP3A4 time-dependent inhibition. Chemical Research in Toxicology, 37(4), 549-560. ↑
- Burns, J., Zalte, A., & Green, W. (2025). Descriptor-based foundation models for molecular property prediction. arXiv preprint arXiv:2506.15792. ↑
- Ash, J. R., Wognum, C., Rodríguez-Pérez, R., Aldeghi, M., Cheng, A. C., Clevert, D. A., ... & Walters, W. P. (2025). Practically significant method comparison protocols for machine learning in small molecule drug discovery. Journal of Chemical Information and Modeling, 65(18), 9398-9411. ↑