Pregnane X Receptor PDB Structure Rerefinement
Improving 66 Pregnane X Receptor structures from the Protein Data Bank using modern refinement protocols to enhance ligand model quality.
Introduction
High-resolution X-ray crystal structures serve as definitive benchmarks for evaluating co-folding methods such as AlphaFold3, Boltz-2, and OpenFold3, particularly for assessing their ability to recover native ligand poses. While these models offer a unified prediction pass, crystallographic data frequently reveal where they struggle, such as maintaining correct stereochemistry or avoiding unphysical atom-atom clashes, areas where traditional docking can sometimes remain more robust. Beyond validation, recent work [1] has shown that target-specific structures can be used to fine-tune models such as OpenFold3, thereby correcting systematic pose errors and moving predictions toward experimentally plausible geometries. Furthermore, X-ray data provide essential geometric parameters—such as site-specific hydrogen-bonding distances and receptor-based excluded volumes that define docking constraints, effectively narrowing the search space and improving the sampling of bioactive conformations. This iterative refinement helps co-folding architectures bridge the gap between statistically probable patterns and the biochemically accurate induced-fit conformational changes observed in X-ray data.
Structures from the PDB are often taken and used directly for model training and evaluation. However, it is worth doing additional processing to improve model quality and curation. While experimental data quality is fixed at the time of deposition, the software used to process and model this data has improved significantly over the past few decades. This means that many structures, older structures in particular, may not reflect the best possible interpretation of the underlying diffraction data. Ligands are particularly difficult to model given their chemical diversity.
Rerefinement, taking existing PDB models and experimental data and rerunning modern refinement protocols, offers a straightforward route to improving structural quality. Here, I describe an effort to rerefine the 66 existing PXR-ligand structures in the PDB and assess its effect on model quality, particularly for ligands.
Refinement Protocols and Model Selection
Structures were refined using two protocols, which I'll refer to as refinement_1 and refinement_2. Refinement_1 used Phenix 1.21 with in-distribution restraints by default, falling back to eLBOW-generated restraints when in-distribution restraints were unavailable for a given ligand. The second, refinement_2, used Phenix 2.0 with the same prioritization, but substituting MOPAC-optimized eLBOW restraints in cases where in-distribution restraints were not available.
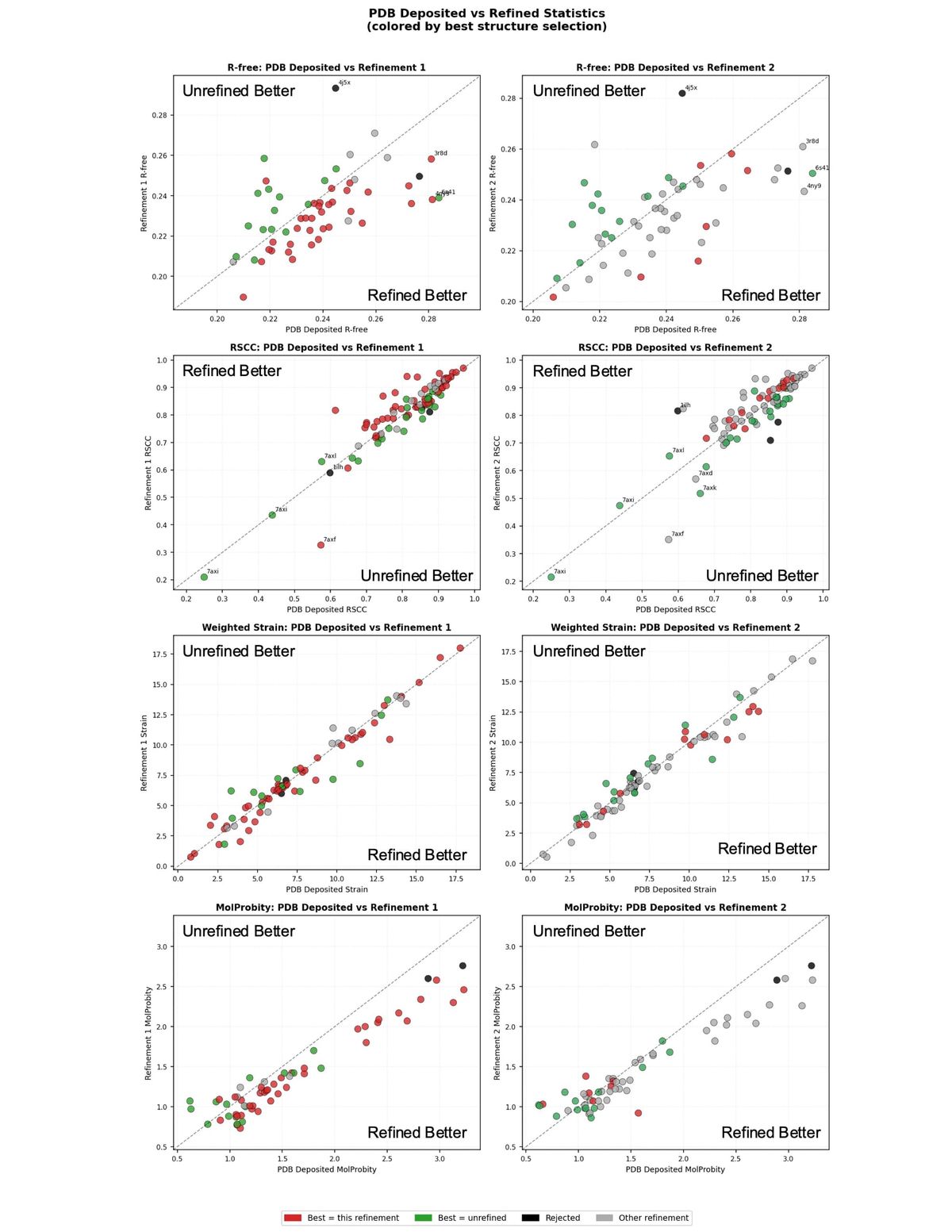
To build the final rerefined dataset, the best version of each structure: unrefined, refinement_1, or refinement_2, was selected. This required balancing several metrics. Since the primary goal is ligand quality, real-space correlation coefficient (RSCC) of the ligand and torsional strain were considered most heavily. RSCC measures how well the refined ligand model agrees with the experimental electron density map, while strain captures whether the ligand geometry is physically reasonable. Rfree, a global measure of model quality, was considered next. MolProbity scores, which assess overall protein geometry, were given the least priority. Importantly, a model that regressed in one statistic could still be selected if sufficiently compelling improvements were made in other statistics and visual inspection. It is worth noting that strain calculations failed for roughly 10% of ligands; for those cases, selection relied on the remaining criteria (Strain was calculated using the method described by [2]
Generally, refinement_1 tended to produce better global statistics, particularly in terms of Rfree. However, refinement_2 produced better models for some, particularly newer depositions with complex ligands.
Results
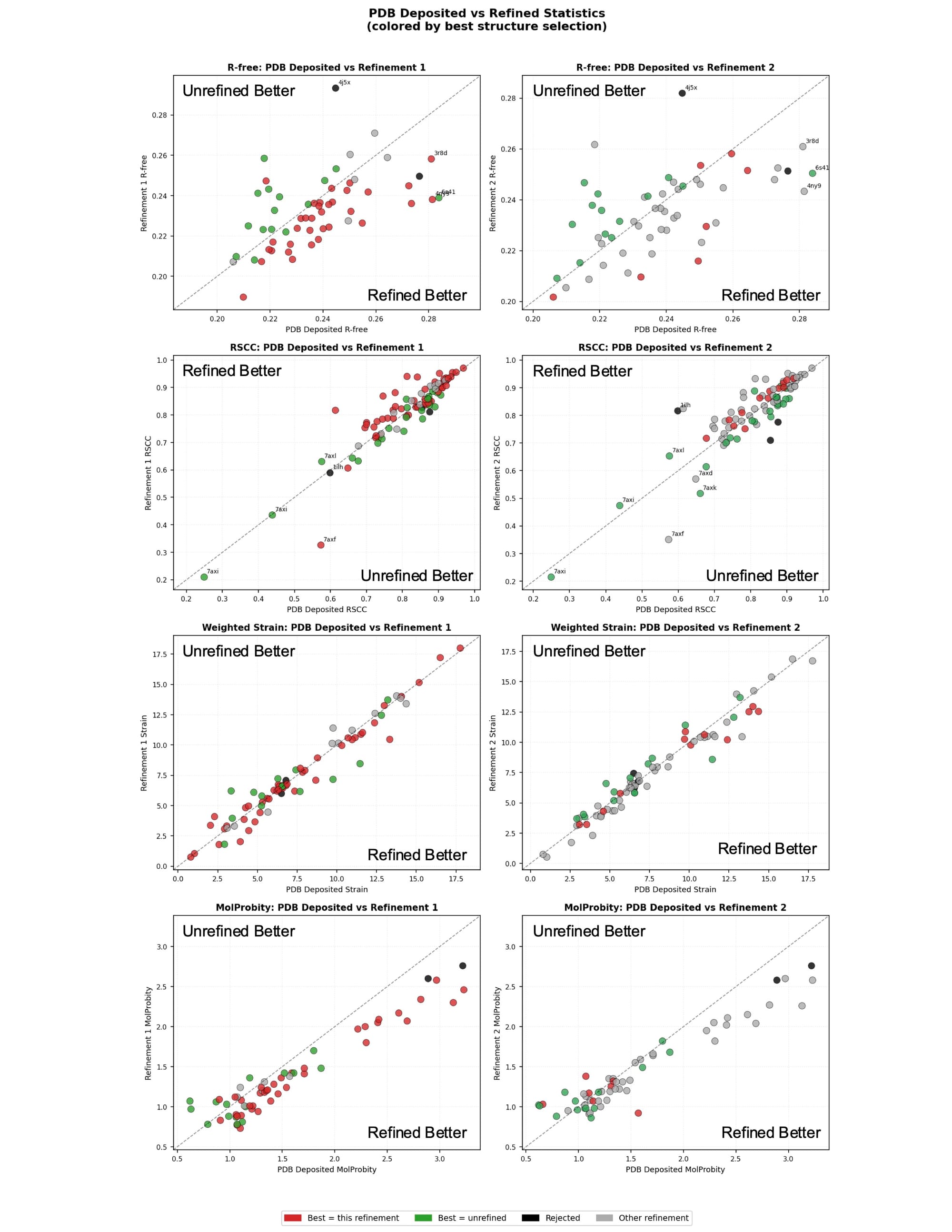
Rerefinement improved model quality modestly across the dataset on average (Figure 1). The majority of structures saw improvements in Rfree. Ligands with an unrefined RSCC below 0.8 tended to improve while ligands with an unrefined RSCC above 0.8 had a roughly even split of improvements and regressions. Ligand strain stayed about the same on average with modest improvements for some ligands. Molprobity improved most significantly. This is not surprising, given that Phenix more directly optimizes protein geometry towards the metrics Molprobity weighs most heavily than other refinement engines. Structures refined with other refinement engines can often see significant Molprobity improvements. A handful of mostly older structures with high Molprobity’s see very substantial Molprobity improvements.
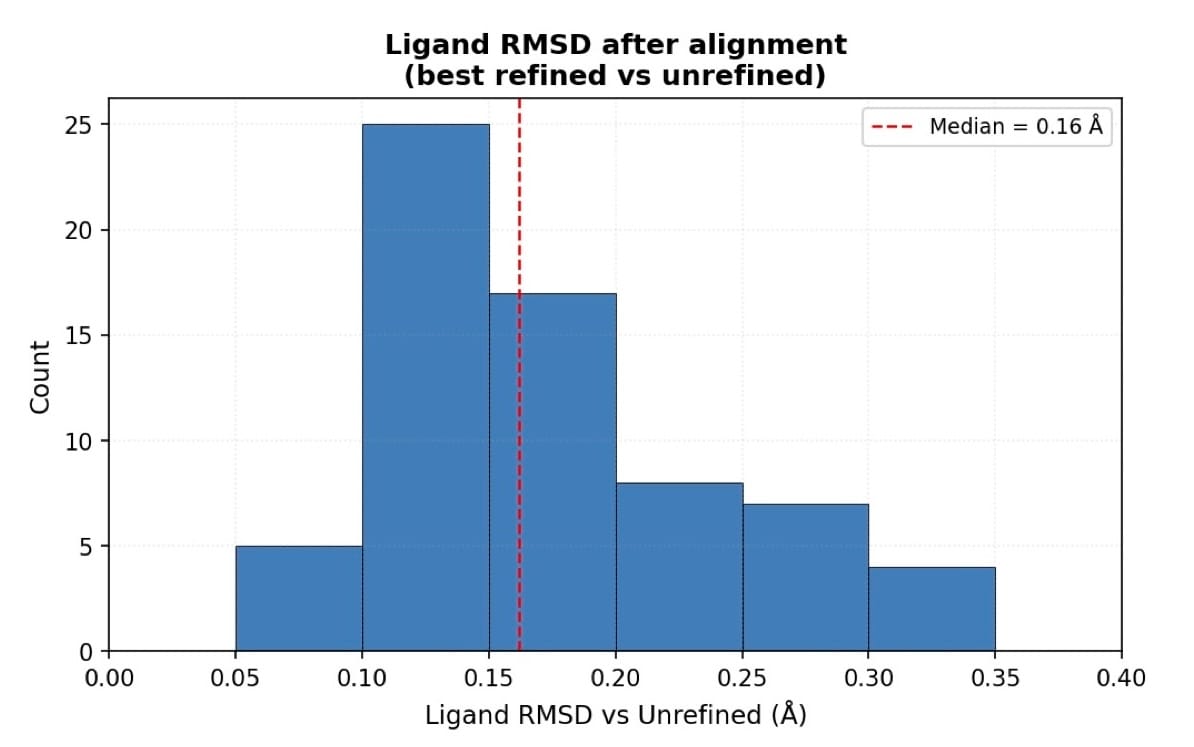
Rerefinement should not result in significant ligand movements. Overall, the RMSD between refined and unrefined ligands is small (Figure 2). The effects of rerefinement are mostly small geometry and density fit improvements.
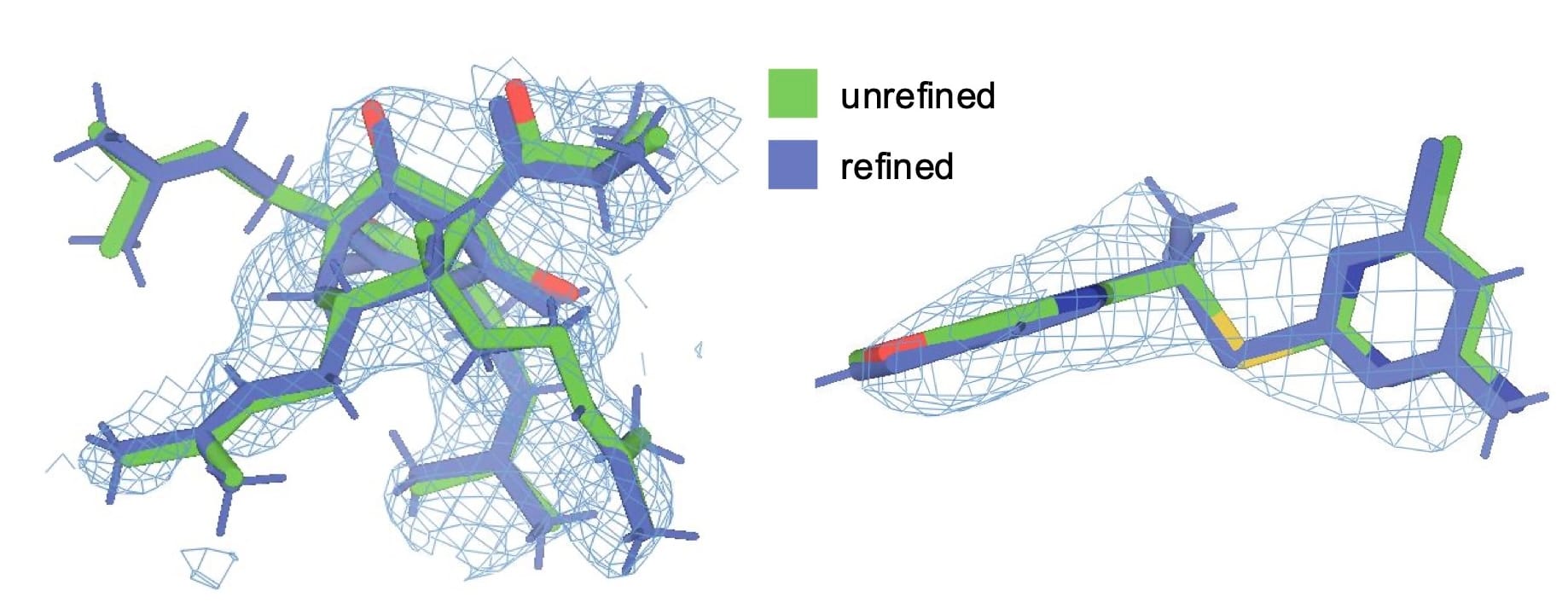
Figure 3 illustrates the visible improvement in ligand density fit for two models. For the ligand in 1m13, RSCC improved from 0.70 to 0.77, and for 3r8d the improvement was more substantial, going from 0.61 to 0.82. In both cases, visual inspection confirms that the refined model fits the electron density more convincingly, with atoms generally more centered in the density.
Notes on Specific Structures
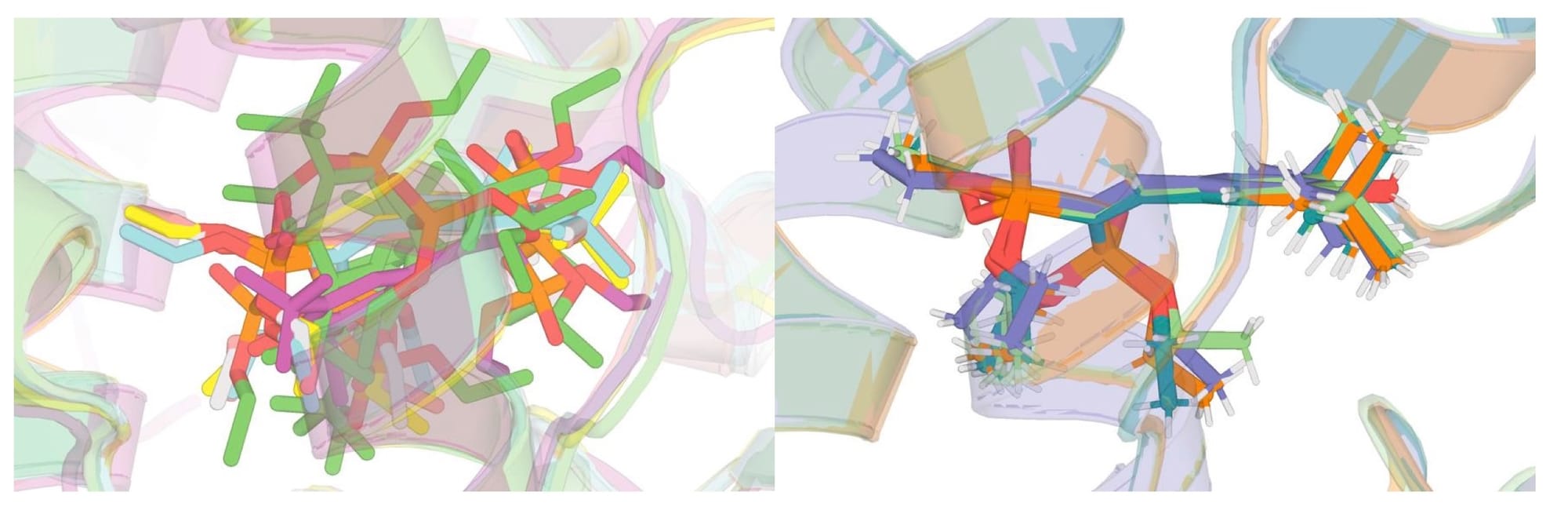
Two structures, 4j5x and 1ilh, were ultimately excluded from the rerefined dataset. Both contain the ligand SR12813, which also appears in 1nrl, 3hvl, 9fzi, and 9fzj. However, 4j5x and 1ilh adopt a noticeably different binding pose from the rest of the group, and are of lower overall quality. Attempts to manually remodel the ligand into a pose consistent with the other structures still yielded a meaningfully different conformation, so these entries were excluded. The rerefined dataset contains a vast increase in agreement of ligand pose across structures compared to the existing PDB dataset (Figure 4).
5 structures 7axd, 7axf, 7axk, 7axl, and 7axi show very poor RSCC values by the numbers, but visual inspection suggests this is an artifact rather than a genuine modeling failure. All five structures contain multiple ligands in close proximity, which can confound RSCC calculations. The effect is particularly pronounced for 7axi, which contains overlapping enantiomers in the binding site. These structures look reasonable when viewed in context of their density, and I do not believe the low RSCC values reflect poor ligand placement. This is illustrative of the difficulty of judging structures purely on metrics.
Notes on Refinement
Generating appropriate ligand restraints is nontrivial. Early runs using basic eLBOW restraints optimization produced poor geometries for macrocycles and fused ring systems, for example hyperforin and rifampicin. These scaffold types are notoriously difficult to parameterize with simple geometry-based methods.
Phenix's in-distribution restraints handled these cases much better. The catch is that in-distribution restraints are not available for all ligands, and coverage drops off noticeably for more recent depositions (approximately for PDB accession codes of 7 or higher). For this reason, refinement_2 was run with eLBOW with mopac optimization, a more robust semi-empirical qm optimization method. For example, 8cf9’s ligand produced an unphysical geometry in refinement_1 but a reasonable geometry in refinement_2 (Figure 5). Interestingly though, refinement with Phenix 2 tended to produce worse metrics than Phenix 1.21, so the majority of selected refined structures were refined with Phenix 1.21
Conclusions
Rerefinement of PDB structures with modern protocols is a tractable and worthwhile exercise for improving ligand model quality, particularly for older depositions. In addition to optimizing ligand and protein coordinates, rerefinement and evaluation can filter out structures that are unrealistic. The choice of restraint generation strategy matters, especially for structurally complex ligands where simple geometry-based parameterization can fail. These findings suggest that the field would benefit from more complex preprocessing and evaluation pipelines, rather than treating PDB depositions as fixed ground truth.
The full rerefined dataset and the refinement scripts can be found here: https://github.com/OpenADMET/pxr_xtal_re-refinement.
References
[2] Shuo Gu, Matthew S. Smith, Ying Yang, John J. Irwin, and Brian K. Shoichet. Journal of Chemical Information and Modeling 2021 61 (9), 4331-4341. DOI: 10.1021/acs.jcim.1c00368