
Lessons Learned from the OpenADMET-ExpansionRx Blind Challenge: Can We Trust Zero-Shot ADMET Predictions?
Maria Castellanos Hugo MacDermott-Opeskin
It’s been more than a month since the OpenADMET-ExpansionRx challenge wrapped up, but the conversation is just getting started. Launched on October 27, 2025, the challenge saw hundreds of participants pushing the boundaries of ADMET endpoint prediction. Seeing this level of engagement, we are more committed than ever to ensuring our platform remains a hub for lively scientific exchange.
As part of this commitment, we recently announced our next blind challenge, focusing on PXR induction, which is set to kick off this April. However, we believe there is still significant value to be extracted from the findings of our recent ExpansionRx study.
In our previous blog post, we discussed key lessons from the wide range of methodologies used by top performers and delved into the performance metrics for each endpoint. Today, we are following up with a deeper dive into a critical question for drug discovery programs: How do public "zero-shot" models actually compare to project-specific models when faced with novel chemical space?
In this context, "zero-shot" inference refers to using a model to predict properties for a new dataset without any additional training, fine-tuning, or exposure to that specific chemical series. Here, we use this as a practical test for a model’s ability to achieve molecular out-of-distribution (MOOD) generalization [1]. By evaluating models on the ExpansionRx dataset, we are measuring how well they translate what they learned from public data to the significant covariate shifts (or changes in the distribution of molecular features and representations) encountered in completely new chemical space.
Is “Zero-shot” inference a good move for ADMET prediction on unseen data?
This is the question that motivated our analysis for this post. We want to thank Anthony Gitter from the University of Wisconsin-Madison for reaching out and suggesting this topic. (You can also find Anthony’s challenge submission, which utilized an AI-scientist approach here).
The universe of publicly available ADMET assays keeps growing every day. For example, ChEMBL, the largest repository of bioactive molecules, contains over 2.88 million distinct compounds and more than 20 million bioactivity data points [2]. At the same time, continuous advancement in machine learning and deep learning architectures is also ongoing. As a motivating example, the recently released Chemprop v.2. [3], a popular open-source framework for molecular property prediction, was the architecture of choice for several of the top performers in our recent blind challenge.
One might assume we are at the stage where models trained on curated public repositories can be used at least somewhat reliably for inference on unseen datasets. Moreover, as "end-to-end" AI agents emerge that depend on these pre-trained tools to score compounds, it becomes increasingly important to ask: can these models truly generalize?
As we will see in this post, the answer is not so simple.
Exploring popular web-based prediction tools
The ecosystem of ADMET-prediction web tools has expanded significantly in recent years. Well-known platforms such as SwissADME [4], ADMETboost [5], and ProTox-II [6] have paved the way for more recent, specialized tools like ADMETlab 3.0 [7] and ADMET-AI [8]. To test the reality of "zero-shot" performance, we focused on the latter two, given their well-maintained web servers and broad endpoint coverage.
ADMET-AI implements a multi-task Chemprop v.1. graph neural network (GNN) architecture enhanced with RDKit molecular features [9]. It is trained on data curated from the Therapeutics Data Commons (TDC) and offers both a web-based tool for up to 1,000 molecules and a Python API for larger datasets.
ADMETlab 3.0 similarly leverages a multi-task Chemprop v.1. model with RDKit features [6]. This platform is trained on over 400,000 molecules across 77 ADMET-related endpoints, curated from massive public repositories including ChEMBL, PubChem, and OCHEM. The web-based platform is available here.
Additionally, we compared these against internal OpenADMET models built using Chemprop v.2. on a pre-trained CheMeleon foundation model. By using an architecture similar to that of the top challenge performers, we can better determine whether performance gaps are due to the model architecture or the training data itself. The model for HLM CLint is available on Hugging Face, while the models for Caco-2 Papp A→B, LogD, and MPPB will be released soon.
Comparing the performance of four selected endpoints
To evaluate the prospective power of these established tools, we compared the per-endpoint performance of ADMET-AI, ADMETlab 3.0 and the OpenADMET model (trained on ChEMBL data) against two baselines from the recent OpenADMET-ExpansionRx challenge:
- A Light Gradient Boosting Machine (LGBM) model utilizing RDKit descriptors [9] trained by the OpenADMET team.
- The winning model submitted by Pebble (Inductive Bio) using their proprietary models and data.
Importantly, the two challenge baselines were trained on the ExpansionRx training split and evaluated on the blinded test split. In contrast, the three web-based tools were applied to the test split in a true "zero-shot" fashion, i.e., without any exposure to the ExpansionRx training data.
ADMET prediction models are trained on curated versions of popular public datasets
While ADMET-AI and ADMETlab 3.0 are both trained on large, chemically diverse datasets, the range and distribution of those values often differ from those in specific, real-world drug discovery campaigns like the ExpansionRx dataset. Before assessing model accuracy, we compared the endpoint distributions of the public training sets (TDC, as curated for ADMET-AI [8] and ChEMBL) against our ExpansionRx train and test splits.
From this point forward, we focus on four primary ADMET endpoints: LogD, Caco-2 Permeability (Papp A→B), HLM CLint, and MPPB. The remaining five endpoints in the ExpansionRx dataset were either unavailable on these web platforms or could not be confidently used for direct comparison (e.g., solubility was reported as LogS by the tools, and it is not clear whether it corresponds to the solubility endpoint in the ExpansionRx dataset, KSOL). Furthermore, while ADMETlab 3.0 reports LogD, ADMET-AI does not, and MPPB is only reported by our internal models.
The distributions for these four endpoints across the ChEMBL, TDC, ExpansionRx Train, and Test split are presented in the figure below.

Overall, the per-endpoint distributions and dynamic range are notably similar. Both ChEMBL and TDC provide ample coverage for these four endpoints, though the ExpansionRx set appears to offer slightly more diversity in its extreme values.
However, aligning these datasets required significant caution. Before we look at the results, it is important to discuss the “Data Harmonization” steps required to make valid comparisons between predictions from different web-based models.
WARNING: The hidden complexity of comparing pre-trained model performance
Comparing external inference models to external datasets is rarely "plug-and-play". For instance, having matching units is critical to evaluating model performance and must not be skipped, especially when using external models. Before evaluating the performance of the web models, we address some issues encountered during data curation and processing.
-
Documentation gap
Although it is standard to perform unit conversions before training or inference, many web-based models do not clearly state the units (or even the scale!) of the resulting values. In this regard, ADMET-AI provided excellent documentation that listed all endpoints, their corresponding units, and links to additional information. In contrast, ADMETlab 3.0 did not provide significant per-endpoint information either on its website or in the source manuscript. This lack of transparency is unfortunately very common in the field and highly problematic for rigorous model comparison. -
Always double-check the units!
Even when units are provided, they could be misleading. For instance, ADMET-AI specified the units Caco-2 A→B permeability to be log(10-6 cm/s). However, a simple analysis of endpoint distribution revealed the values were actually in log(cm/s). This conversion closely matched the ChEMBL distribution, whereas the stated units did not. We strongly advise the reader to personally verify the distribution of endpoints, even if units are provided in advance. -
Human vs mouse benchmarks
A subtle but critical element to consider is the endpoint species. While the ExpansionRx dataset provides Mouse PPB data, both ADMET-AI and ADMETlab 3.0 predict Human PPB. It is tempting to use these values as proxies for one another, especially when a model doesn't explicitly label the species in the output. However, plasma protein binding can vary significantly across species due to differences in protein sequences, binding affinities, and concentrations. This is the reason this data is collected across species in the first place! We strongly advise the reader to verify the target species of the training data before comparing it with another model. Using models trained on human data to predict mouse endpoints, or vice versa, will likely introduce significant interspecies bias that cannot be corrected by simple unit conversion. Consequently, we excluded the PPB analysis for the web-based models and focused the analysis on our internal model, which was specifically trained on MPPB data. -
Intrinsic clearance vs. plasma clearance.
The HLM CLint endpoint required the most intensive pre-analysis. As we discussed in our previous blog post, the Well-Stirred Model provides the necessary link between raw enzymatic rates and total hepatic clearance (CLH):
Where QH is hepatic blood flow (assumed at 20 mL/min/kg), and fu is the fraction of unbound drug.
The ADMET-AI model predicts intrinsic clearance in vitro (µL/min/mg), so we applied the standard scaling factors presented in our blogpost to convert these into in vivo clearance (mL/min/kg) for comparison. However, ADMETlab 3.0 is significantly more complicated to evaluate, as it predicts clearance as plasma (total) clearance. To get these values into a comparable format, we had to calculate an estimate of the intrinsic in vivo clearance by using the inverse form of the Well-Stirred equation:
By using the predicted values for unbound drug in plasma (HPPM) and assuming a human hepatic blood flow constant (QH) of 20 mL/min/kg, we were able to approximate the intrinsic hepatic clearance predicted by the model.
For prospective prediction, however, we strongly recommend against following this workflow. Using this inverse approximation to the Well-Stirred model is mathematically "dangerous" for three reasons: 1) Error Amplification: The equation significantly amplifies errors in CLH as the measured clearance approaches the QH constant (the denominator approaches zero). 2) Compounded Uncertainty: Incorporating predicted values for fu further multiplies the potential prediction error. And 3) Sensitivity to Strong Binders: The fu factor makes the result highly sensitive, such that a tiny difference in unbound fraction (e.g., 0.001 vs 0.002) can double the calculated clearance.
Consequently, we adopt this approximation solely for the purposes of this retrospective analysis and recommend that our readers stick to models trained directly on CLint data whenever possible.
Zero-shot public models do not extrapolate to unseen datasets like the ExpansionRx set
Even after all this work matching units and physiological scales to give these tools the best possible chance at a fair comparison, the results were disappointing.
If the endpoint distributions are so similar between the public benchmarks and our specialized dataset, why can’t we directly apply these models to the ExpansionRx dataset? To answer this, we looked at the raw predictive power of ADMET-AI, ADMETlab 3.0 and our internal models in a true "zero-shot" setting. The regression plots below show the experimental versus predicted values for our blinded test split, paired by endpoint.
- LogD: We observed a stark performance gap for this endpoint. While ADMETlab 3.0 showed poor alignment with experimental values (R2 = 0.02, MAE = 0.82 and Spearman ⍴ = 0.6), our internal models achieved significantly better results with an R2 of 0.50, a MAE of 0.57, and a Spearman ⍴ of 0.70. This disparity is particularly unexpected given the abundance of public data for LogD, suggesting that the "zero-shot" failure of web tools may be a matter of implementation rather than a lack of available training data.

- Caco-2 (A→B) Permeability: While Caco-2 Papp showed a smaller Mean Absolute Error (MAE) than LogD, the predictive accuracy was even worse. This resulted in a negative R2 for all zero-shot models studied, including our internal model. Passive permeability is typically considered a tractable task because of its strong correlation with lipophilicity. However, these global models could not match the performance seen in the challenge, where Pebble achieved an R2 of 0.62.

- HLM CLint: Intrinsic clearance (CLint) was the most challenging task for challenge participants, both in terms of relative error and ranking. CLint is notoriously difficult to predict because it is highly sensitive to small structural changes (activity cliffs), making it hard for models to generalize across different chemical series. With negative R2 values, both ADMET-AI and ADMETlab 3.0 proved incapable of meaningfully predicting clearance in this "zero-shot" context. While ADMETLab 3.0 showed the highest deviation, possibly due to the plasma-to-intrinsic clearance conversion performed, the results suggest that neither of these tools is currently suited for zero-shot clearance prediction on the ExpansionRx dataset.

- MPPB: While this endpoint was only available in our internal models, it showed the best relative performance across all endpoints, aligning with our challenge observations. This is likely due to the smaller dynamic range of this endpoint within the test set, as well as its strong underlying correlation with molecular size and lipophilicity.
Limitations in endpoint coverage in public datasets make it difficult to do “zero-shot” predictions on novel datasets
To quantify the translation gap between models trained on public data vs those trained on the ExpansionRx dataset, we compared the predictive performance of the zero-shot web tools against two models that had the advantage of having been trained directly on the ExpansionRx training set: our LGBM baseline and the winning model of the challenge from Pebble (Inductive Bio).
The plot below shows the distribution of performance, measured by R2 and Mean Absolute Error (MAE), across 1,000 bootstrapped samples for each of the four endpoints and four models (on the y-axis). Bootstrapping was used rather than the more rigorous cross validation procedure recommended in this paper [10], as it was not possible to cross validate web-based models.

The difference in performance is remarkable. The winning model from Pebble performed best across all four endpoints, followed closely by the LGBM baseline. In contrast, both ADMET-AI and ADMETlab 3.0 showed consistently poor performance. To confirm the robustness of these observations, we performed a Tukey HSD test on the bootstrapped distributions. The resulting Compact Letter Display (CLD) showed that all models belong to distinct statistical groups, confirming that performance gaps between the models are statistically significant.
In some cases, our internally trained models trained on public data (available on Hugging Face) closely matched or even surpassed the baseline trained on ExpansionRx data (specifically for LogD and MPPB). For these endpoints, one could argue that either the abundance of public data allows state-of-the-art models to generalize, as is the case with LogD, or the endpoint benefits from high-quality data in a closely related task, as seen with MPPB.
-
The results for LogD provide perhaps the most interesting insight into the "data availability vs. model" debate. While Pebble’s model was the only one to approach the intrinsic error limit of 0.2–0.3 MAE, the performance of our internal model (trained on public ChEMBL data) highlights an important trend. Our internal model achieved an MAE of ~0.56, performing nearly on par with the locally-trained LGBM baseline. This suggests that for LogD, the sheer volume of public data enables a global model with a robust architecture to perform as well as or better than a simple model trained on the specific program data.
However, the fact that our model and the LGBM baseline both stalled around 0.5, while Pebble reached <0.3, hints that the remaining gap may not be fully explained by data availability, but instead lies in the specifics of the model, featurization, or ensembling used by top challenge performers. This improved performance by Pebble could also be attributed to the use of proprietary data.
In sharp contrast, ADMETlab 3.0 performed significantly worse than both the public and local models. This is a surprising result given the vast amount of LogD7.4 data available in public repositories, and it highlights that even with "solved" endpoints, not all web-based implementations are created equal.
-
HLM CLint proved to be one of the most difficult endpoints to learn. ADMETlab 3.0 underperformed significantly here, likely due to the physiological scaling issues and documentation gaps we identified earlier. A similar poor performance was observed in Caco-2 permeability.
-
MPPB was the "easiest" endpoint to learn, largely due to its small dynamic range. Here, the performance of the zero-shot models was much closer to the supervised LGBM model. These patterns align with the challenge results.
The lack of overlap with the chemical space of public datasets may result in poor model performance
To investigate why the zero-shot models struggled, we analyzed chemical similarity across three key datasets: ChEMBL (the source for our OpenADMET models and the primary source for ADMETlab 3.0), TDC (curated for ADMET-AI), and our ExpansionRx dataset.
Using the RDKit, we calculated Morgan Fingerprints (radius 2, 2048 bits) and compared nearest-neighbor Tanimoto similarities within each dataset (left panel). The ExpansionRx set shows high internal similarity, as expected for molecules derived from specific discovery campaigns. In contrast, the TDC datasets used as training for the zero shot models show a much broader distribution, reflecting their curation for wide chemical space coverage.
In the right panel, we show the inter-dataset similarity. While similarity is moderately high between the ExpansionRx train and test splits, it is significantly lower between the test split and the public ChEMBL and TDC datasets. This is likely the primary driver behind the poor performance: models trained on ChEMBL or TDC are essentially operating in a different region of chemical space and are unable to extrapolate to a domain they haven’t "seen."
Strikingly, the average nearest-neighbor similarity between the ExpansionRx test set and ChEMBL is below 0.4, and for the curated TDC dataset, it drops below 0.3. In cheminformatics, the "95% noise level" for bit-based Morgan2 fingerprints is approximately 0.27 [9]. Below this threshold, any retrieved compound is statistically as likely to be a random molecule as it is to be "related" to the query. We can conclude, therefore, that a significant fraction of the molecules in these public datasets provides little to no actionable information for the model to learn the structure-activity relation (SAR) of our unseen test set. The models are forced to extrapolate into entirely unseen territory rather than interpolate within a known domain.

This hypothesis becomes even clearer when visualizing the chemical space for each endpoint via GTM plots.
- LogD: The chemical spaces of the train and test sets are similar. ChEMBL’s space is broad enough to overlap with our test set, which explains why our internal ChEMBL-trained model performs decently on this endpoint.
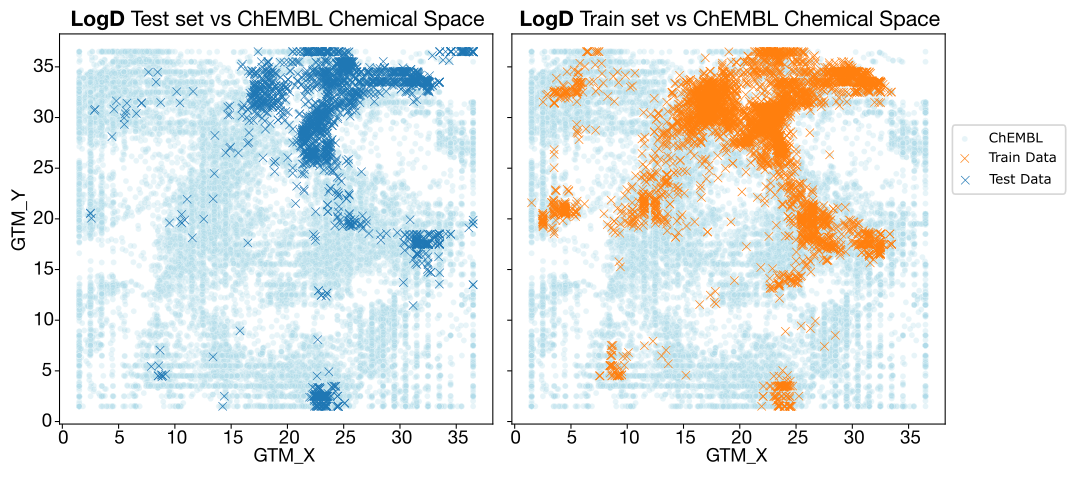
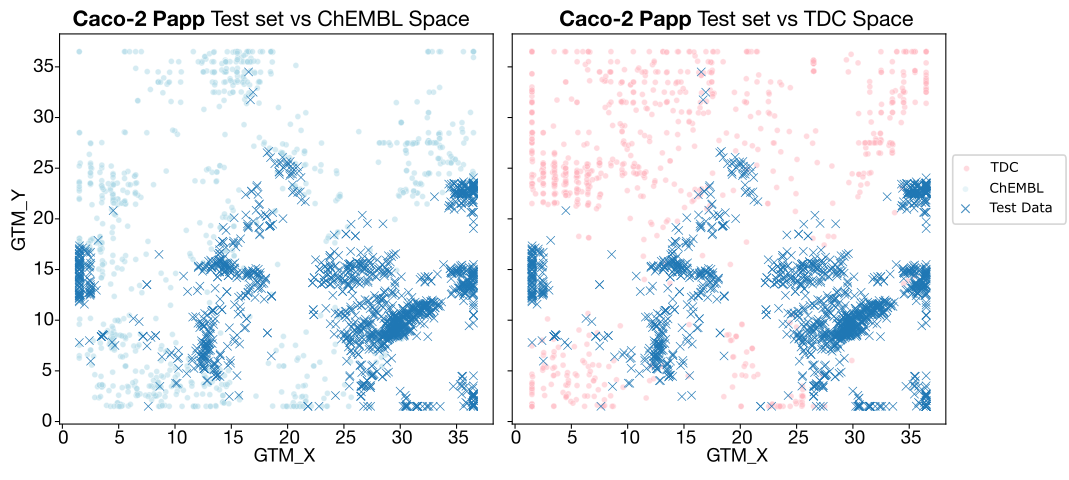
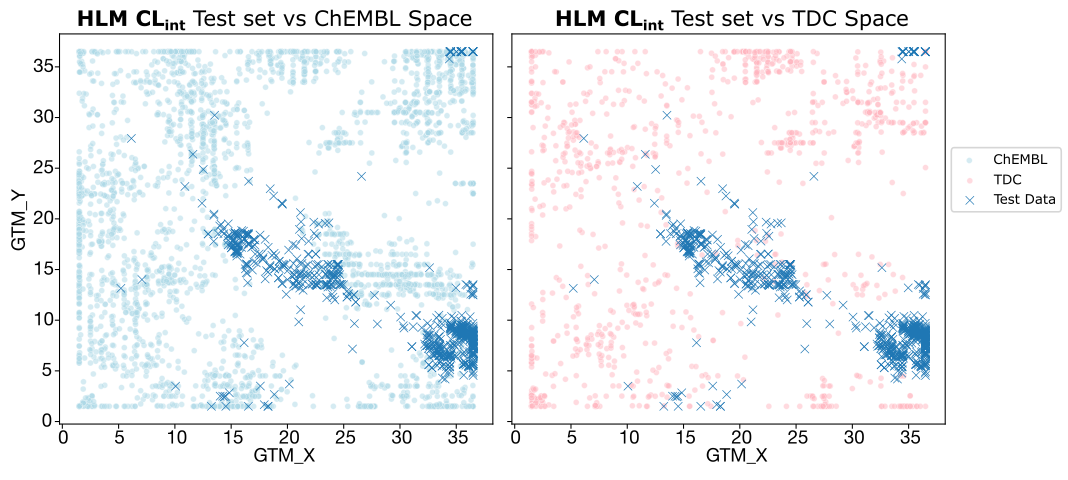
- Caco-2 Papp and HLM CLint: Overlap is extremely poor, especially for TDC. Since ADMET-AI relies solely on TDC, it lacks the "chemical context" necessary to make accurate predictions for these series. Intrinsic clearance is further complicated by the fact that it is a notoriously difficult endpoint to learn, even with good data.
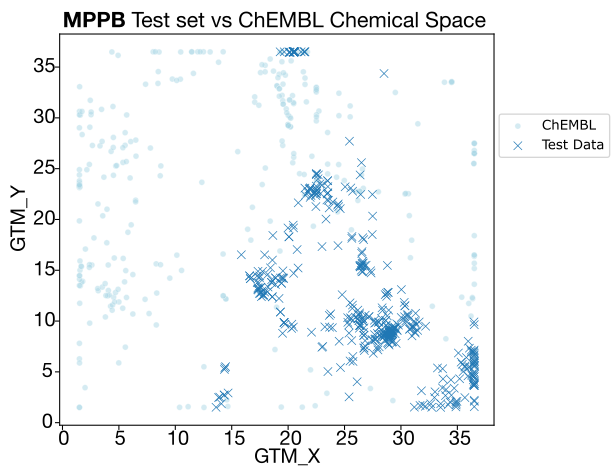
- MPPB: While overlap with ChEMBL is also limited, the better-than-expected zero-shot results are likely due to the narrow dynamic range of the endpoint itself, making "lucky" guesses more statistically probable.
So, now what?
While web-based tools are a useful guide for a compound’s ADMET properties, they are not yet at a stage where they can be used to confidently predict performance in entirely novel chemical space. The takeaway is clear: Do not rely on these models for "zero-shot" ADMET predictions in active drug discovery programs.
Our analysis of the current landscape reveals a stark divide between what is possible today and what still needs work:
- Successful local interpolation: The blind challenge demonstrates that with current public data and modeling strategies, we can successfully predict endpoints within a project (e.g., using earlier compounds to predict later ones). In these cases, the model is interpolating within a known chemical domain.
- The failure of zero-shot extrapolation: For almost every critical ADMET parameter, public models fail to generalize to unseen datasets due to a lack of necessary chemical context.
LogD stands as the important exception here, where zero shot models may show some utility. This endpoint is considered relatively “easy to learn” due to its additive nature. Our in-house zero-shot model trained on public data was shown to be competitive with a baseline trained on project data in this work. While still far behind state of the art models trained on project data (e.g. Pebble), reasonable performance compared to a baseline suggests that the high volumes of data for this endpoint on public repositories, combined with the learnability of LogD, helps models generalize more readily. This seems to also benefit correlated endpoints, such as MPPB.
However, it is important to note that reaching "data saturation" may not be as easy for all endpoints. For more complex biological endpoints, such as clearance and cell permeability, significantly more data, and perhaps more sophisticated biological descriptors, could be needed to overcome the inherent biological noise and "activity cliffs" that characterize these tasks.
This is where defining a model's Applicability Domain (AD) will become critical. The challenge we present here applies to any model performing inference on out-of-distribution data. If we can accurately quantify whether a new molecule sits within or outside a model's AD, we can decide whether or not to trust that prediction—we're currently preparing a blog post on this topic, so stay-tuned!
Ultimately, LogD proves the goal is reachable, but our quest remains the same: we need more high-quality, high-density public data. Initiatives like the OpenADMET-ExpansionRx Challenge are crucial for releasing the kind of rich datasets that will enable us to build models we can use with confidence in the future.
As a reminder, please indicate whether you want to be included as a co-author, and fill out your name and affiliation on this feedback form, as you want it to appear in our future publication. Importantly, even if you have already provided your contact information with your submission, please re-enter it in the survey so we have a centralized database.
Questions or Ideas?
We’d love to hear from you! Whether you want to learn more, have ideas for future challenges, or wish to contribute data to our efforts.
Join the OpenADMET Discord or contact us at openadmet@omsf.io.
Let’s work together to transform ADMET modeling and accelerate drug discovery!
Acknowledgements
We gratefully acknowledge Jon Ainsley, Andrew Good, Elyse Bourque, Lakshminarayana Vogeti, Renato Skerlj, Tiansheng Wang, and Mark Ledeboer for generously providing the Expansion Therapeutics dataset used in this challenge as an in-kind contribution.
References
- Tossou, P., Wognum, C., Craig, M., Mary, H., & Noutahi, E. (2024). Real-world molecular out-of-distribution: Specification and investigation. Journal of Chemical Information and Modeling, 64(3), 697-711.
- Zdrazil, B., Felix, E., Hunter, F., Manners, E. J., Blackshaw, J., Corbett, S., ... & Leach, A. R. (2024). The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic acids research, 52(D1), D1180-D1192.
- Graff, D. E., Morgan, N. K., Burns, J. W., Doner, A. C., Li, B., Li, S. C., ... & Greenman, K. P. (2025). Chemprop v2: An Efficient, Modular Machine Learning Package for Chemical Property Prediction. Journal of Chemical Information and Modeling.
- Daina, A., Michielin, O., & Zoete, V. (2017). SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific reports, 7(1), 42717.
- Tian, H., Ketkar, R., & Tao, P. (2022). ADMETboost: a web server for accurate ADMET prediction. Journal of molecular modeling, 28(12), 408.
- Banerjee, P., Eckert, A. O., Schrey, A. K., & Preissner, R. (2018). ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic acids research, 46(W1), W257-W263.
- Fu, L., Shi, S., Yi, J., Wang, N., He, Y., Wu, Z., ... & Cao, D. (2024). ADMETlab 3.0: an updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic acids research, 52(W1), W422-W431.
- Swanson, K., Walther, P., Leitz, J., Mukherjee, S., Wu, J. C., Shivnaraine, R. V., & Zou, J. (2024). ADMET-AI: a machine learning ADMET platform for evaluation of large-scale chemical libraries. Bioinformatics, 40(7), btae416.
- Landrum, G. (2021). Fingerprint similarity thresholds for database searches.
- Ash, J. R., Wognum, C., Rodríguez-Pérez, R., Aldeghi, M., Cheng, A. C., Clevert, D. A., ... & Walters, W. P. (2025). Practically significant method comparison protocols for machine learning in small molecule drug discovery. Journal of chemical information and modeling, 65(18), 9398-9411.
