Cofolding? I hardly know her! - Cofolding Methods on ADMET Targets
Part 1 of a series on cofolding methods for ADMET targets: using structure prediction to model protein–ligand complexes for key Avoidome anti-targets like PXR and CYP3A4.

Late-stage drug development failures are often not due to poor on-target binding, but to liabilities such as poor absorption, restricted tissue distribution, rapid metabolism, inadequate excretion, or unforeseen toxicity (together, ADMET). At OpenADMET, we aim to address this by providing open access to predictive models and high-quality data for ADMET targets, leveraging structural biology and structure-based design to interrogate the AVOIDOME, the collection of proteins to avoid binding during drug design.
As part of our efforts at OpenADMET, we are carrying out X-ray crystallography and cryo-EM studies on core AVOIDOME targets. Our initial efforts are focused on two clinically important anti-targets in human drug metabolism: the pregnane X receptor (PXR) and Cytochrome P450 3A4 (CYP3A4). PXR is a nuclear receptor and xenobiotic sensor that acts as a master regulator of drug metabolism, inducing key enzymes including the cytochrome P450 family. CYP3A4 is a main member of this group, responsible for metabolizing half of known therapeutic drugs.
Structure-based drug discovery depends on knowing how a ligand binds. Historically, that meant waiting for a validated experimental structure, such as through X-ray crystallography or cryo-EM. The 2021 release of AlphaFold2 changed that, demonstrating that protein structures could be predicted with high accuracy from sequence alone, winning the 14th Critical Assessment of Protein Structure Prediction (CASP14) challenge by a huge margin, achieving a median backbone RMSD of 0.96Å, significantly outperforming the other competing computational methods. In its wake, AlphaFold3 and other cofolding methods capable of predicting protein-ligand complex structures emerged. Boltz-2 and OpenFold3 are among the most capable open-source implementations of this approach, and their accessibility makes them particularly relevant to efforts like ours.
A lot of hype has been cultivated around the buzzword “cofolding”. The excitement is warranted, as the scientific progress is real. However, recent publications have reported a clear relationship between training set similarity and cofolding performance, raising important questions about how these methods perform on targets underrepresented in their training data. Questions remain about whether the accuracy achieved for protein structure prediction extends to protein-ligand complexes. Our results for PXR and CYP3A4 speak directly to this concern. These findings highlight critical performance gaps that we hope will guide developers in refining their cofolding approaches for exactly these kinds of challenging systems. Before deploying any of these methods in real drug discovery workflows, it is important to understand their limitations. At OpenADMET, we are not in the business of building these methods. We are in the business of breaking them, finding where they fail, how far they can be pushed, and being honest with the community about what they can and cannot do.
Our approach
We evaluated cofolding performance on existing Protein Data Bank (PDB) structures for PXR and CYP3A4, generating predictions with Boltz-2 and OpenFold3 (OF3). This should be an easy bar to clear as the majority of these structures are in the training set of both Boltz-2 and OF3. As a sanity check, we also evaluated a small subset of p38 structures from the FEP+ benchmark, as this is a highly-studied target as well as a part of the four-target test set which Boltz-2 performed well on.
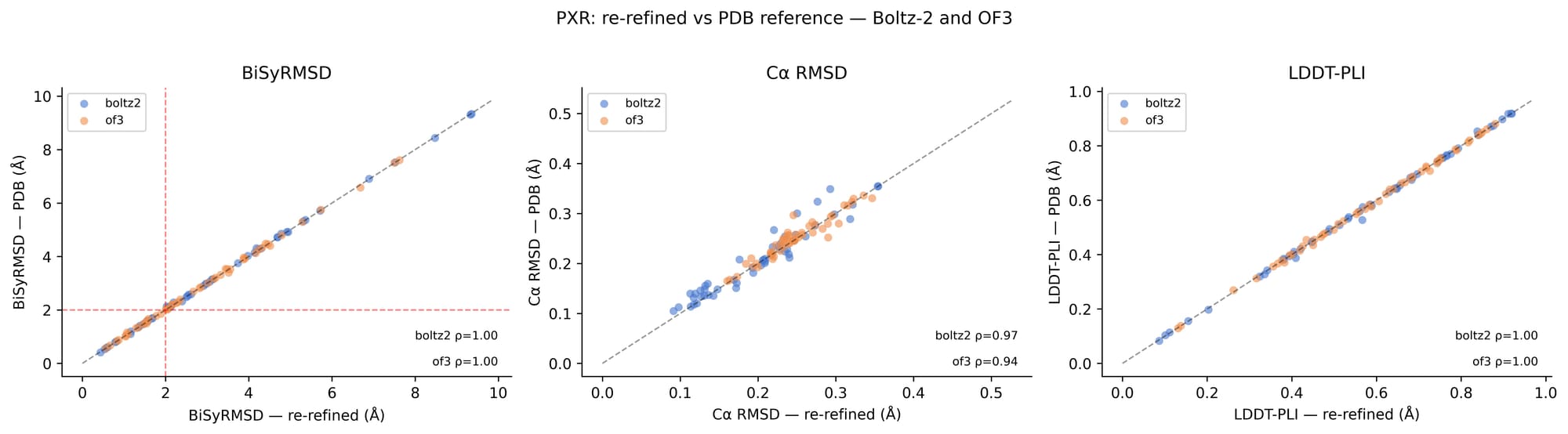
Five samples per structure were generated for each method, allowing us to assess the consistency of predictions and to select the sample with the best ligand placement for evaluation, rather than relying on a single prediction. Prior to analysis, we filtered out multi-ligand structures, to avoid ambiguity in single-ligand pose evaluation, and any structures containing organometallic compounds, as their coordination chemistry was not handled well by the scoring metric pipeline. For PXR, a subset of structures, for which we believe a better interpretation of the underlying diffraction data was possible, were re-refined using Phenix to improve density fit and geometry, though we note that this re-refinement did not significantly change our results (see Figure S1). The re-refinement effort itself is a subject of one of our blog posts. Check it out here.
For our evaluation we used the oracle ranking approach, using the PDB structures as ground-truth, rather than utilizing the confidence ranking scores output by the methods. This represents an optimistic upper bound on performance. In practice, without access to experimental structures, users would rely on the models’ confidence ranking, which, as we later note, provides little reliable signal.
Predictions were assessed using four complementary structural metrics, summarized in the table below. The sample with the best ligand root mean square deviation (RMSD) was selected as the representative prediction for each structure, on which the Binding-Site Superposed, Symmetry-Corrected Pose RMSD (BiSyRMSD) and the Local Distance Difference Test for Protein-Ligand Interactions (LDDT-PLI) were then computed.
The backbone is not the problem.
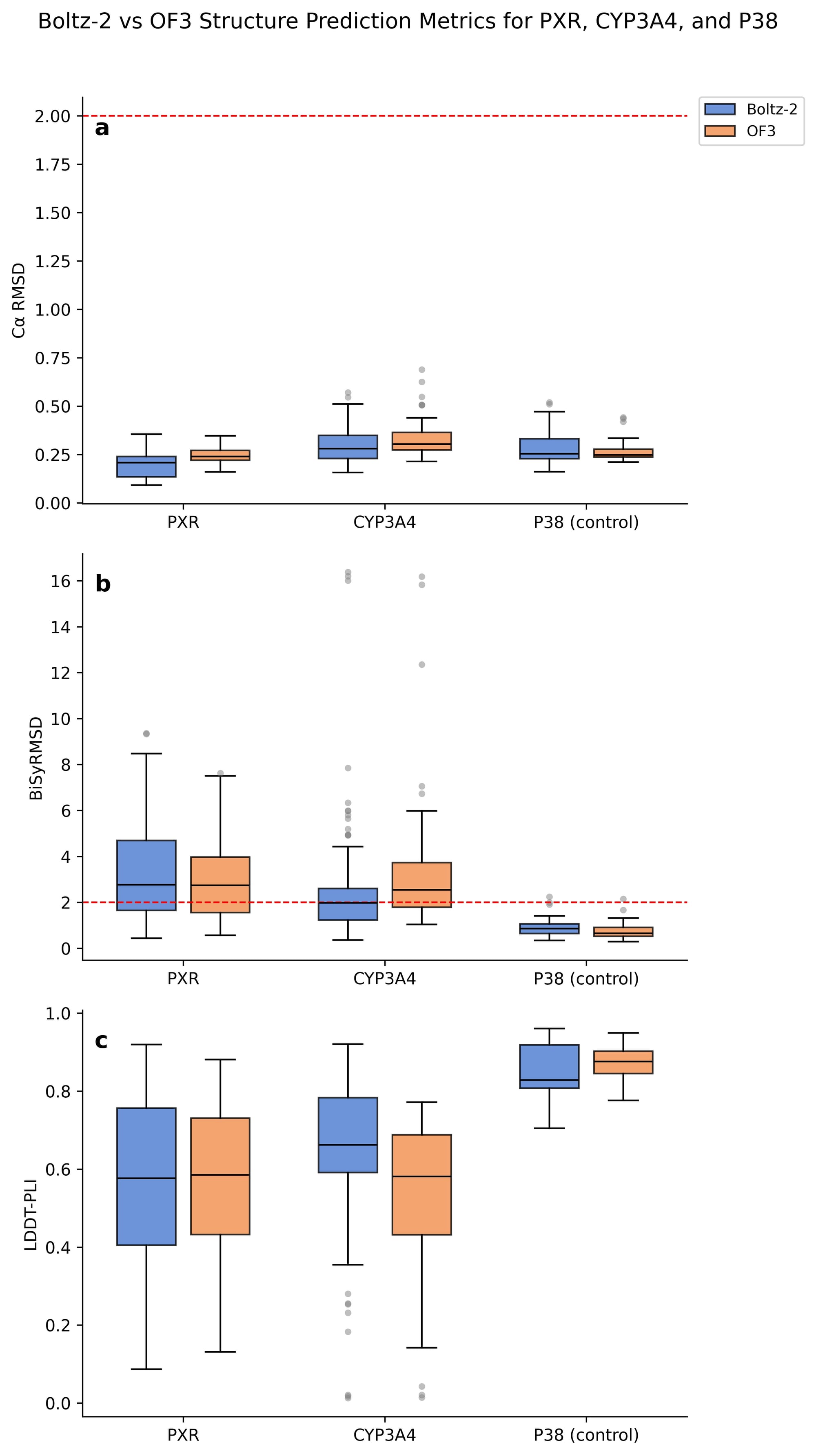
Figure 1a shows that for PXR, CYP3A4, and p38, backbone accuracy is strong across both Boltz-2 and OF3, with no C⍺ RMSD exceeding 0.75Å. The ligand positioning, however, was not reproduced as consistently.
Using a BiSyRMSD threshold of < 2Å as the criterion for a successful prediction, Boltz-2 correctly places the ligand in only 38.5% of PXR structures and 57.8% of CYP3A4 structures. OF3 succeeds in 32.7% and 31.5%, respectively (Figure 1b). Overall, PXR shows a higher median BiSyRMSD than CYP3A4 across both methods, despite CYP3A4 having outliers reaching as high as 16Å. We will explore these outliers later in the post.
These failures are not simply a matter of the ligand being in the wrong position. In Figure 1c we see that LDDT-PLI and BiSyRMSD are negatively correlated: structures with poor ligand placement also are not reproducing protein-ligand interaction distances. So we are finding that the ligand is not only in the wrong pose, but these structures are missing the key interactions that define the binding site.
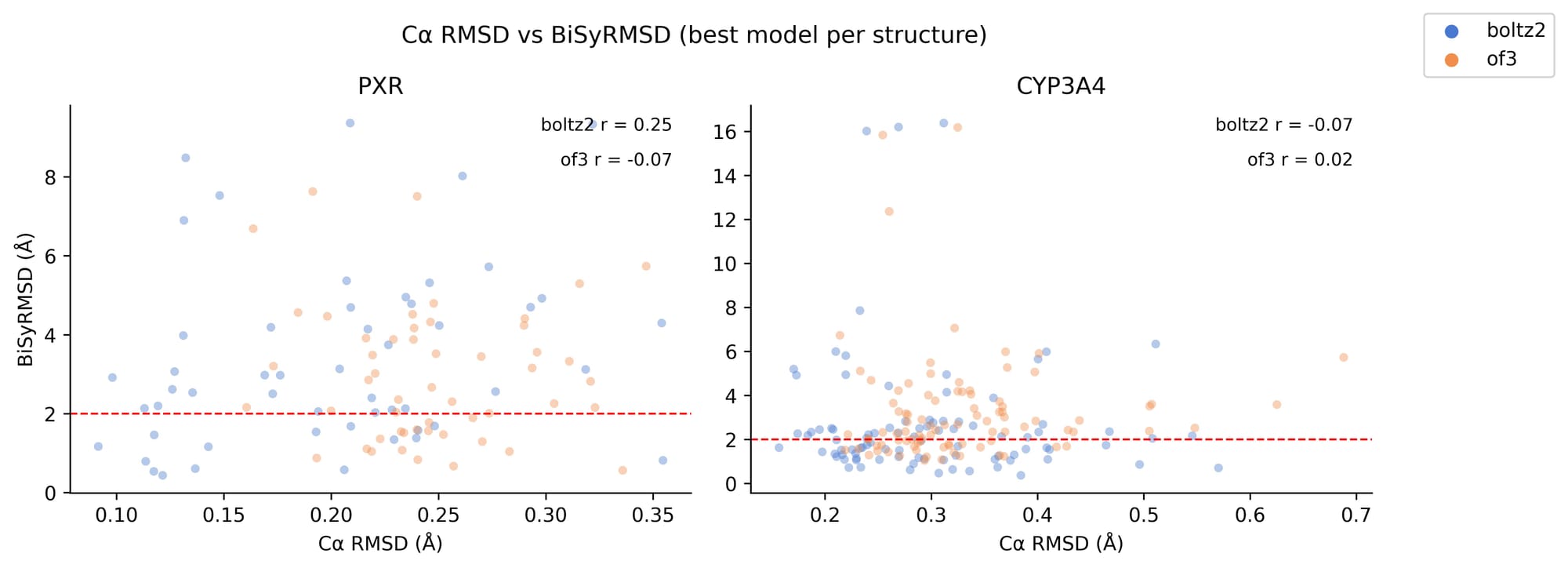
Backbone accuracy offers no predictive signal for ligand pose quality either. C⍺ RMSD does not correlate with ligand RMSD across either target (Figure 2). The decoupling is particularly striking for CYP3A4, where some of the lowest C⍺ RMSD values coincide with the highest BiSyRMSD values. Accurate global protein structure prediction is therefore not a sufficient condition for accurate ligand pose prediction.
For our baseline, p38, we see high performance across all three metrics for both Boltz-2 and OF3. This is unsurprising as p38 is a well-characterized system across the PDB. Most BiSyRMSD values sit comfortably below the 2Å threshold, C⍺ RMSD is low and consistent, and LDDT-PLI scores are high for both methods, indicating that the predicted protein-ligand interactions closely match the experimental structures. Against this high performing baseline, failures for our two prototypical ADMET targets are quite obvious.
Already in training. Still wrong.
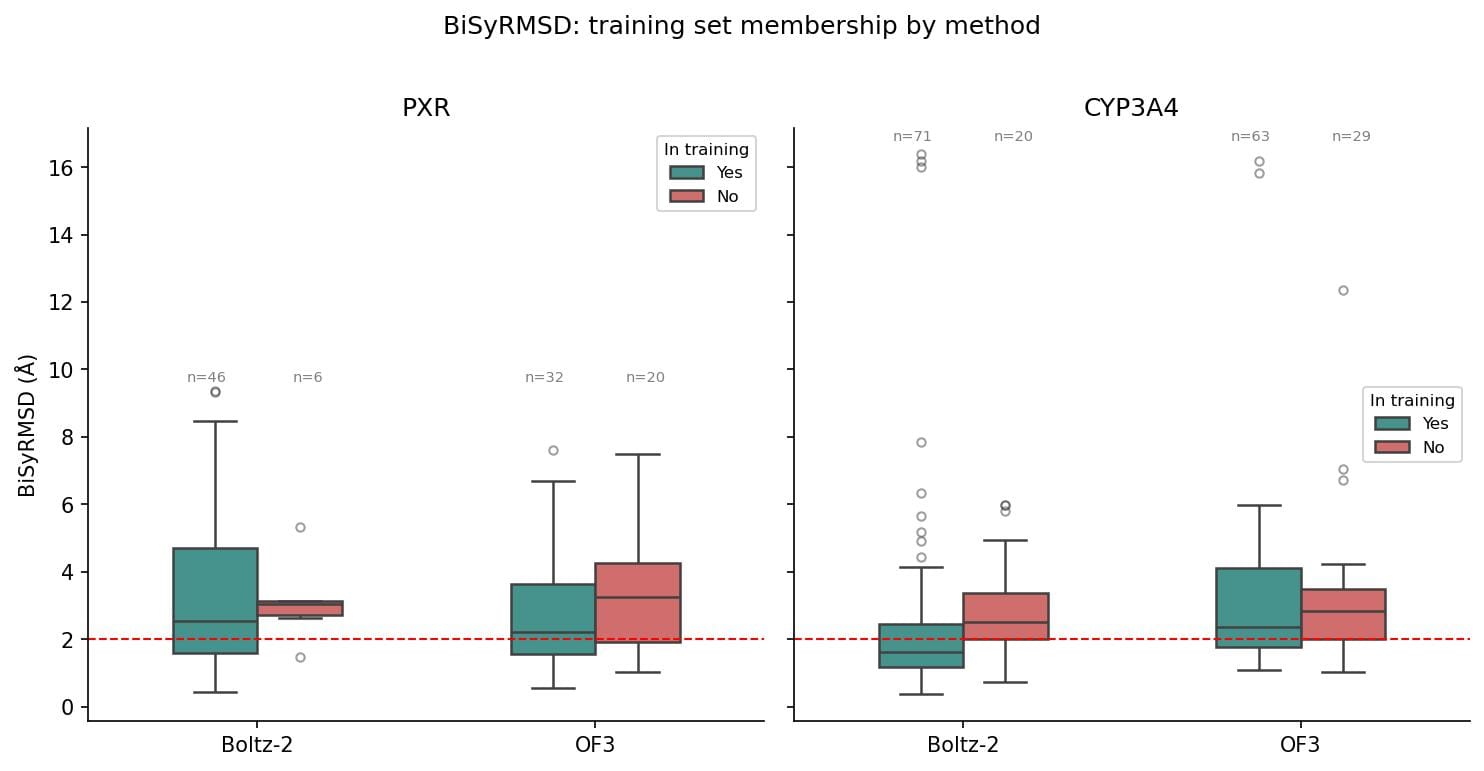
It is worth noting that the majority of tested structures fall within the training sets of both Boltz-2 and OF3. For PXR, 88.5% were present in the Boltz-2 training set (cutoff June 2023), while 61.5% were in the training set for OF3 (cutoff September 2021). Of the tested structures for CYP3A4, 78.9% were included in the Boltz-2 training set and 68.5% were in that of OF3. Figure 3 shows the BiSyRMSD performance as a function of training set membership for each method. Performance is consistent on all structures, whether they are present in the training set or not.
The implications here are hard to ignore. These are not novel structures the models have never encountered. And yet, even on structures within their training data, both methods fail on ligand pose prediction, while continuing to reproduce the protein backbone well. This suggests the models are not learning ligand chemistry from the structures they have seen. Kinases, for example, account for thousands of PDB entries and are among the most well studied protein families in structural biology. The performance of Boltz-2 and OF3 on p38 reflects that representation. PXR and CYP3A4 are not as well represented and the performance gap supports this. These models may be converging toward some average over the training distribution, but for ADMET targets that are underrepresented relative to the rest of the PDB, that average is not a useful one. The underlying reason is still an open question, but regardless of the cause, we cannot currently trust these models to place the ligand correctly, even for structures they have seen before.
What wrong looks like.
We have demonstrated that both methods fail to correctly place the ligand in the majority of evaluated structures for both targets. So what do these predictions actually look like?
Figure 4 shows four structure prediction examples, two for each method and target. The caption for each structure indicates the PDB ID, the color of the experimental structure within the visualization, and the corresponding lowest BiSyRMSD value. Looking at these examples, a pattern emerges that is representative of failures throughout our results: ligands often land in the right binding pocket, but with the wrong geometry or in the wrong orientation (often both).
Let’s look at PXR first. For 6XP9 (Figure 4a), the predicted poses cluster correctly in the ligand binding domain but are at completely the wrong orientation relative to the experimental structure. For 8SVX (Figure 4b), every predicted sample shows an approximate 180° flip.
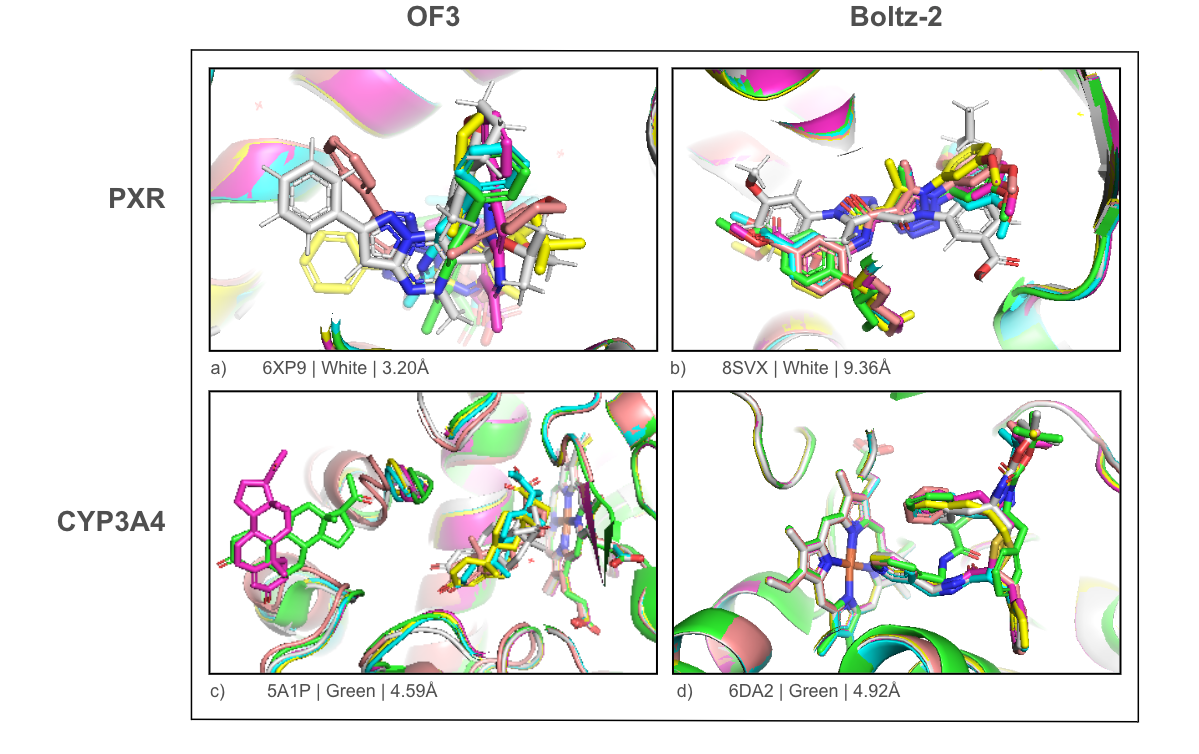
CYP3A4 results are a bit more interesting. As shown above, the median BiSyRMSD is lower for CYP3A4 than PXR, even with the extreme outliers. It appears the heme may act as a geometric anchor. The models get the heme placement right every time, providing a fixed point in space to orient the ligand. In 6DA2 (Figure 4d), all predicted samples coordinate with the heme iron. The placement is seemingly better constrained than in PXR, but the orientation is still off. When diving deeper into the extreme outliers for CYP3A4 (BiSyRMSD > 10Å), we find these are all misplaced allosteric ligands binding in the orthosteric site, in this case specifically progesterone. In 5A1P (Figure 4c), the vast majority of predictions place the ligand near the heme rather than the peripheral allosteric pocket. OF3 finds this pocket once, at a BiSyRMSD of 4.59Å.
Every structure shown here has a best-case BiSyRMSD above 2Å. Some are closer than others, but none cross the threshold for a successful prediction.
It is also worth noting that the confidence and ranking scores output by these methods offer no useful signal; they are highly compressed, ranging from 0.92 to 0.93 for Boltz-2 and 0.91 to 0.96 for OF3, show no meaningful correlation with BiSyRMSD, and rarely rank the best predicted structure first.
Summary
Across both PXR and CYP3A4, cofolding methods reproduce protein backbone structures well, while poorly reproducing ligand pose. Neither Boltz-2 nor OpenFold3 reliably place ligands for the ADMET targets correctly, even for structures within their training sets. For these targets, the predicted protein-ligand interactions do not reflect the chemistry of the binding site, even when the ligand is placed in the right general region. Whether this reflects a fundamental limitation of the approach, or simply the underrepresentation of ADMET targets in training data is an open question, but one worth asking. If these methods are going to be used in practice, getting the protein right is not enough.
Next Steps
The failures we observe are systematic, and we think training data is at least part of the story. ADMET targets like PXR and CYP3A4 are underrepresented in the PDB relative to kinases and other well-studied drug targets, and that imbalance likely shapes what these models have learned. As fine-tuning pipelines are established for these methods, we will examine whether targeted tuning can improve performance. If the failures are data-driven, fine-tuning on new data should help. Our colleagues at UCSF are actively generating high quality experimental structure data for ADMET targets, which will provide exactly the kind of target-specific training data needed to test this hypothesis.
Stay tuned for Part 2 of this series! While this post focuses on structure prediction, its counterpart will evaluate the affinity prediction capabilities of these methods; specifically, the Boltz-2 affinity head and AQAffinity for OF3.
Supplementary Information: